
Nội dung toàn văn Tiêu chuẩn quốc gia TCVN 6134:2009 (EPA Method 8321A) về Chất lượng đất – Phương pháp sắc ký lỏng hiệu năng cao/nhiệt phun/ khối phổ (HPLC/TS/MS) hoặc Detector cực tím (UV) để xác định hợp chất không bay hơi có thể chiết trong dung môi
TIÊU CHUẨN QUỐC GIA
TCVN 6134 : 2009
CHẤT LƯỢNG ĐẤT – PHƯƠNG PHÁP SẮC KÝ LỎNG HIỆU NĂNG CAO/NHIỆT PHUN /KHỐI PHỔ (HPLC/TS/MS) HOẶC DETECTOR CỰC TÍM (UV) ĐỂ XÁC ĐỊNH HỢP CHẤT KHÔNG BAY HƠI CÓ THỂ CHIẾT TRONG DUNG MÔI
Soil quality – Solvent extractable nonvolatile compounds by high performance liquid chromatography/thermospray/mass spectrometry (HPLC/TS/MS) or ultraviolet (UV) detection
Lời nói đầu
TCVN 6134 : 2009 thay thế cho TCVN 6134 : 1996.
TCVN 6134 : 2009 hoàn toàn tương đương với Method 8321A của Cơ quan bảo vệ môi trường Hoa Kỳ (EPA Method 8321A).
TCVN 6134 : 2009 do Ban kĩ thuật tiêu chuẩn quốc gia TCVN/TC 190 Chất lượng đất biên soạn, Tổng cục Tiêu chuẩn Đo lường Chất lượng đề nghị. Bộ Khoa học và Công nghệ công bố.
CHẤT LƯỢNG ĐẤT – PHƯƠNG PHÁP SẮC KÝ LỎNG HIỆU NĂNG CAO/NHIỆT PHUN /KHỐI PHỔ (HPLC/TS/MS) HOẶC DETECTOR CỰC TÍM (UV) ĐỂ XÁC ĐỊNH HỢP CHẤT KHÔNG BAY HƠI CÓ THỂ CHIẾT TRONG DUNG MÔI
Soil quality – Solvent extractable nonvolatile compounds by high performance liquid chromatography/thermospray/mass spectrometry (HPLC/TS/MS) or ultraviolet (UV) detection
1. Phạm vi và áp dụng
1.1 Phương pháp này đề cập đến việc sử dụng sắc ký lỏng hiệu năng cao (HPLC) cặp khối phổ-nhiệt phun (TS-MS) và/hoặc tia cực tím (UV) để xác định thuốc nhuộm azo, hợp chất phospho hữu cơ và tris(2.3-dibromopropyl) phosphat, hợp chất axit phenoxy clo hóa và các este của chúng, cacbamat trong nước thải, nước ngầm và nền mẫu đất/trầm tích. Các dữ liệu phân tích tro bay của các loại thuốc trừ cỏ axit phenoxy clo hóa cũng được trình bày (Bảng 15), tuy nhiên độ thu hồi của phần lớn các hợp chất là rất thấp nên có thể thấy các phân tích dùng quy trình chiết xuất theo phương pháp này sẽ có hiệu quả thấp. Phương pháp này có thể áp dụng cho các hợp chất không bay hơi khác mà có thể chiết bằng dung môi và phân tích bằng HPLC, và có thể ion hóa dưới điều kiện nhiệt phun để xác định khối phổ hoặc có thể được xác định bằng detector UV. Có thể xác định được các hợp chất dưới đây bằng phương pháp này.
|
Tên hợp chất |
Số CASa |
|
Thuốc nhuộm azo |
|
|
Disperse Red 1 |
2872-52-8 |
|
Disperse Red 5 |
3769-57-1 |
|
Disperse Red 13 |
126038-78 6 |
|
Disperse Yellow 5 |
6439-53-8 |
|
Disperse Orange 3 |
730-40-5 |
|
Disperse Orange 30 |
5261-31-4 |
|
Disperse Brown 1 |
17464-91-4 |
|
Disperse Red 3 |
6535-42-8 |
|
Disperse Red 23 |
85-86-9 |
|
Thuốc nhuộm Anthraquinon |
|
|
Disperse Blue 3 |
2475-46-9 |
|
Disperse Blue 14 |
2475-44-7 |
|
Disperse Red 60 |
17418-58-5 |
|
Thuốc nhuộm comarin (hợp chất benzopyrone) |
|
|
Hợp chất làm sáng vải |
|
|
Hợp chất làm sáng vải 61 |
8066-05-5 |
|
Hợp chất làm sáng vải 236 |
3333-62-8 |
|
Hợp chất Alkaloid |
|
|
Caffein |
58-08-2 |
|
Strichnin |
57-24-9 |
|
Hợp chất phospho hữu cơ |
|
|
Methomyl |
16752-77-5 |
|
Thiofanox |
39796-18-4 |
|
Famphur |
52-85-7 |
|
Asulam |
3337-71-1 |
|
Diclorvos |
62-73-7 |
|
Dimethoat |
60-51-5 |
|
Disulfoton |
298-04-4 |
|
Fensulfothion |
115-90-2 |
|
Merhos |
150-50-5 |
|
Methyl parathion |
298-00-0 |
|
Monocroctophos |
919-44-8 |
|
Nled |
300-76-5 |
|
Phorat |
298-02-2 |
|
Trichlorfon |
52-68-6 |
|
Tris(2.3-dibromopropyl) phosphat (tris-BP) |
126-72-7 |
|
Hợp chất axit phenoxy clo hóa |
|
|
Dalapon |
75-99-0 |
|
Dicamba |
1918-00-9 |
|
2,4-D |
94-75-7 |
|
MCPA |
94-74-6 |
|
MCPP |
7085-19-0 |
|
Dichforprop |
120-36-5 |
|
2,4,5-T |
93-76-5 |
|
Silvex (2,4,5-TP) |
93-72-1 |
|
Dinoseb |
88-85-7 |
|
2,4-DB |
94-82-6 |
|
2,4-D, butoxyethanol ester |
1929-73-3 |
|
2,4-D, ethylthexyl ester |
1928-43-4 |
|
2,4,5-T, butyl ester |
93-79-8 |
|
2,4,5-T, butoxyethanol ester |
2545-59-7 |
|
Cacbamat |
|
|
Aldicarb |
116-06-3 |
|
Adicarb sulfone |
1646-88-4 |
|
Aldicarb sulforxide |
1646-87-3 |
|
Aminocarb |
2032-59-9 |
|
Barban |
101-27-9 |
|
Benomyl |
17804-35-2 |
|
Bromacil |
314-40-9 |
|
Bendiocarb |
22781-23-3 |
|
Carbaryl |
63-25-2 |
|
Carbendazim* |
10605-21-7 |
|
3-Hydroxycarbofuran |
16655-82-6 |
|
Carbofuran* |
1563-66-2 |
|
Chloroxuron |
1982-47-4 |
|
Chloropropham |
101-21-3 |
|
Diuron* |
330-54-1 |
|
Fenuron |
101-42-8 |
|
Fluometuron |
2164-17-2 |
|
Linuron* |
330-55-2 |
|
Methiocarb |
2032-65-7 |
|
Methomyl* |
16752-77-5 |
|
Mexacarbate |
315-18-4 |
|
Monuron |
150-68-5 |
|
Neburon |
555-37-3 |
|
Oxamyl* |
23135-22-0 |
|
Propachlor |
1918-16-7 |
|
Propham |
122-42-9 |
|
Propoxur |
114-26-1 |
|
Siduron |
1982-49-6 |
|
Tebuthiuron |
34014-18-1 |
CHÚ THÍCH
* Số đăng ký hóa chất
* Các chất cacbamat này được thử trong đánh giá liên phòng thí nghiệm: tất cả các chất khác được thử khi đánh giá đơn phòng thí nghiệm.
1.2 Phương pháp này có thể áp dụng để phân tích các hợp chất không bay hơi hoặc nửa bay hơi khác.
1.3 Tris-BP đã được phân loại là chất gây ung thư. Vật liệu chuẩn tinh khiết và dung dịch chuẩn gốc phải được xử lý trong tủ hút.
1.4 Tiêu chuẩn này được thiết kế để phát hiện các hợp chất axit phenoxy clo hoá (dạng axit tự do) và các este của chúng mà không sử dụng bước thủy phân và este hoá trong qui trình chiết, tuy nhiên việc thuỷ phân thành dạng axit sẽ định lượng một cách đơn giản.
1.5 Các hợp chất được chọn để phân tích bằng HPLC/MS là những hợp chất có thành phần rất khó phân tich bằng phương pháp sắc ký truyền thống (ví dụ sắc ký khí). Độ nhạy của phương pháp này phụ thuộc vào mức độ cản trở trong nền mẫu và thay đổi theo các nhóm hợp chất và thậm chí khác nhau giữa các hợp chất trong cùng một nhóm. Ngoài ra, giới hạn phát hiện (LOD) phụ thuộc vào điều kiện vận hành của máy đo khối phổ. Ví dụ, LOD đối với cafein trong chế độ monitoring phản ứng chọn lọc: (SRM) là 45 pg dung dịch chuẩn được bơm (bơm 10 mL), trong khi đối với Disperse Red 1 thì LOD là 180 pg. LOD của cafein trong điều kiện quét mạch tứ cực đơn là 84 pg và 600 pg đối với Disperse Red 1 trong điều kiện quét tương tự.
1.6 Giới hạn xác định từ thực nghiệm (LOD) đối với các chất cần phân tích được trình bày trong Bảng 3, 10, 13 và 14. Để nhận dạng thêm các hợp chất, có thể dùng MS/MS (CAD- phân ly hoạt tính xung) làm phương pháp tùy chọn của phương pháp này.
1.7 Phương pháp này chỉ được sử dụng hoặc được giám sát bởi các nhà phân tích có kinh nghiệm trong việc sử dụng sắc ký lỏng hiệu nâng cao có dùng detector khối phổ hoặc detector UV. Người phân tích cũng cần có kỹ năng diễn giải sắc đổ lỏng và khối phổ. Mỗi người phân tích phải chứng minh được khả năng đưa ra các kết quả có thể chấp nhận được theo phương pháp này.
2. Tóm tắt phương pháp
2.1 Phương pháp này trình bày phương pháp sắc ký lỏng hiệu năng cao pha đảo (RP/HPLC) và điều kiện khối phổ (MS) phun nhiệt (TS) và hoặc điều kiện tia cực tím (UV) để phát hiện các chất cần phân tích. Phân tích định lượng được thực hiện bằng TS/MS, sử dụng phương pháp nội chuẩn hoặc ngoại chuẩn. Dịch chiết mẫu được phân tích bằng cách bơm trực tiếp vào bề mặt phun nhiệt hoặc lên trên bề mặt nhiệt phun sắc ký lỏng. Chương trình rửa giải gradien được dùng trong sắc ký để phân tách các hợp chất. Có thể phát hiện được cả bằng ion hoá ion âm (điện cực phóng điện) và ion hoá ion dương, với khối phổ từ cực đơn. Vì phương pháp này dựa trên kỹ thuật HPLC, nên sử dụng detector tia cực tím (UV) là tùy chọn khi phân tích mẫu thường nhật.
2.2 Trước khi sử dụng phương pháp này, phải sử dụng các kỹ thuật chuẩn bị mẫu phù hợp.
2.2.1 Mẫu để phân tích các hợp chất axit phenoxy clo hoá được chuẩn bị bằng Method 8151 sửa đổi (xem 7 1.2) Nói chung, một lít mẫu nước hoặc 50 g mẫu đất được điều chỉnh pH, chiết bằng dietyl ete, làm cô đặc và đưa về dung môi axetonitril.
2.2.2 Mẫu để phân tích các chất phân tích khác được chuẩn bị bằng kỹ thuật chiết đã có. Nói chung, mẫu nước được chiết ở pH trung tính bằng metylen clorua, dùng Method 3500 phù hợp. Method 3500 thích hợp sử dụng metylen clorua/axeton (1:1) được dùng cho các mẫu rắn. Kỹ thuật vi chiết cũng áp dụng với chiết Tris-BP từ các nền mẫu lỏng và không lỏng.
2.2.3 Đối với cacbamat một lít mẫu nước hoặc 40 gam mẫu chất rắn được chiết bằng metylen clorua (tham khảo Method 3500 thích hợp), cô đặc (nên sử dụng máy cô quay có adapter) và đưa về dung môi metanol.
2.3 Phương pháp khẳng định tuỳ chọn khối phổ-nhiệt phun/khối phổ (TS-MS/MS) cũng được quy định. Việc khẳng định có thể thu được bằng sử dụng MS/MS Phân tách hoạt hoá va chạm (CAD) hoặc dây kỵ nước CAD.
3. Viện dẫn
3.1 Tham khảo Method 3500, 3600, 8000 và 8151.
3.2 Với một số hợp chất trong phương pháp này, sử dụng cột làm sạch dùng cột florisil (Method 3620) đã cho kết quả độ thu hồi nhỏ hơn 85 %, vì vậy không nên áp dụng phương pháp này với tất cả mọi hợp chất. Tham khảo Bảng 2 của Method 3620 về độ thu hồi các hợp chất phospho hữu cơ như là một hàm số của phần chiết florisil.
3.3 Các hợp chất có ái lực proton cao có thể che lấp một số chất cần phân tích, Do vậy HPLC phải được sử dụng như là một bộ tách sắc ký để phân tích định lượng.
3.4 Những khó khăn về phân tích gặp phải với các hợp chất phospho hữu cơ cụ thể khi áp dụng phương pháp này có thể bao gồm như sau (nhưng không phải là tất cả):
3.4.1 Metyl parathion cho thấy một số phân huỷ nhỏ khi phân tích.
3.4.2 Naled có thể bị khử brôm hoá tạo thành dạng dichlovos.
3.4.3 Merphos thường chứa các chất nhiễm bẩn từ oxit merphos. Quá trình oxy hoá merphos có thể xảy ra trong khi lưu giữ và trong quá trình đưa vào máy khối phổ.
Tham khảo Method 8141 về các vấn đề với các hợp chất khác do liên quan đến các phương pháp chiết khác nhau.
3.5 Hợp chất axit phenoxy clo hoá, là axit hữu cơ mạnh, phản ứng ngay với các chất kiềm và có thể mất trong khi phân tích. Do vậy, dụng cụ thuỷ tinh và bông thuỷ tinh phải được rửa bằng axit và natri sunphat phải được axit hoá bằng axit sunfuric trước khi dùng để tránh hiện tượng này.
3.6 Do tính phản ứng của thuốc trừ cỏ clo, các dung dịch chuẩn phải được chuẩn bị trong axetonitril. Quá trình metyl hoá sẽ xảy ra chậm nếu được chuẩn bị trong metanol.
3.7 Benomyl được biết là chất phân huỷ nhanh thành carbendazim trong môi trường (Tài liệu tham khảo 21).
3.8 Dung môi, thuốc thử, dụng cụ thuỷ tinh và một số dụng cụ chuẩn bị mẫu khác có thể ảnh hưởng làm kết quả thu được rời rạc hoặc đường nền cao, hoặc cả hai, dẫn đến diễn giải nhầm sắc đồ hoặc phổ. Tất cả các vật liệu này phải chứng minh được không có chất cản trở trong các điều kiện phân tích bằng cách phân tích thuốc thử trắng. Cần phải lựa chọn thuốc thử và độ tinh khiết của dung môi bằng cách chưng cất trong hệ thống bằng thuỷ tinh.
3.9 Các chất cản trở cùng chiết từ mẫu sẽ thay đổi theo các nguồn. Thời gian lưu của chất phân tích phải được kiểm chứng bằng cách sử dụng các chất chuẩn đối chứng
3.10 Lựa chọn sử dụng phương pháp HPLC/MS/MS nhằm mục đích trong khẳng định chất phân tích cụ thể. Các phương pháp này ít phụ thuộc vào bản chất hoá học hơn là các phương pháp khối phổ khác.
4. Thiết bị và vật liệu
4.1 HPLC/MS
4.1.1 Sắc ký lỏng hiệu năng cao (HPLC): Hệ thống phân tích có hệ phân phối dung môi được lập chương trình và tất cả các phụ kiện yêu cầu bao gồm vòng bơm mẫu (có dung tích vòng tối thiểu 10 mL. cột phân tích, bộ làm sạch khí. v.v… Hệ thống phân phối dung môi tối thiểu phải là hệ thống dung môi đôi. Hệ thống sắc ký phải có khả năng hoạt động trên khối phổ (MS).
4.1.1.1 Bơm bổ sung sau cột HPLC: Có thể dùng một bơm bổ sung sau cột. Bơm này cần phải là một bơm xylanh và không cần phải có chương trình dung môi.
4.1.1.2 Nên dùng các cột HPLC- Cần có cột bảo vệ và cột phân tích.
4.1.1.2.1 Cột bảo vệ – C18 cột bảo vệ pha đảo, 10 mm x 2,6 mm ID thuỷ tinh, 0,5 mm hoặc tương đương.
4.1.1.2.2 Cột phân tích – C18 cột pha nghịch, 100 mm x 2 mm ID. cỡ hạt ODS-hypersil 5 mm; hoặc cột pha đảo C6, 100 mm x 2 mm ID, cỡ hạt MOS2-hypersil 3 mm: hoặc tương đương
4.1.2 Giao diện HPLC/MS
4.1.2.1 Bộ vi trộn – 10 mL, tương thích với hệ thống cột HPLC có hệ thống bổ sung dung môi sau cột.
4.1.2.2 Mặt phân giới: Bề mặt tiếp xúc ion hoá nhiệt phun và nguồn sẽ cho tín hiệu trả lời chuẩn chấp nhận được cho mỗi chất cần phân tích ở nồng độ yêu cầu. Nguồn này phải có khả năng giải phóng ra cả ion dương và ion âm và có điện cực hoặc dãy phóng điện.
4.1.3 Hệ thống khối phổ: Là khối phổ tứ cực đơn có khả năng quét từ 1 amu đến 1000 amu. Máy đo phổ này cũng phải có khả năng quét từ 150 amu đến 450 amu trong 1,5 giây, hoặc ít hơn, sử dụng năng lượng electron 70 von (danh định) trong các trường hợp tương tác eletron dương và âm. Ngoài ra khối phổ phải có khả năng tạo ra phổ khối lượng hiệu chuẩn đối với PEG 400, 600 hoặc 800 (xem 5.14) hoặc các chất khác được dùng làm chất hiệu chuẩn.
4.1.3.1 Khối phổ tứ cực ba tuỳ chọn có khả năng phát ra phổ ion con khi có khí va chạm trong tứ cực thứ hai và vận hành trong chế độ tứ cực đơn.
4.1.4 Hệ thống dữ liệu: Hệ thống máy tính cho phép thu nhận liên tục và lưu giữ trên thiết bị có thể đọc được tất cả phổ khối lượng thu được trong suốt thời gian của chương trình sắc ký và phải tương thích với máy đo khối phổ. Máy tính phải có phần mềm cho phép mọi file dữ liệu MS được tìm kiếm đối với các ion của từng khối lượng xác định và các ion này được vẽ đồ thị theo thời gian hoặc số quét. Loại đồ thị này được định nghĩa là đường ion chiết xuất hiện tai (EICP). Phần mềm cũng phải có khả năng tổng hợp số lượng ion của mỗi EICP trong từng khoảng thời gian hoặc số lần quét nhất định. Phải có phần mềm máy tính vận hành riêng cho từng thiết bị đo khối phổ.
4.2 HPLC có detector UV: Hệ thống phân tích có hệ thống bơm dung môi được lập trình cho ít nhất một hệ dung môi đôi, và tất cả các phụ kiện được yêu cầu bao gồm bơm tiêm, vòng bơm 10 mL, cột phân tích, bộ làm sạch khí. v.v... Bơm tự động là tùy chọn, nhưng phù hợp khi phân tích nhiều mẫu. Cột được qui định trong 4.1.1.2 cũng đươc sử dụng cho hệ thống này.
4.2.1 Nếu detector UV được dùng trong bộ đôi thiết bị phân tích với giao diện nhiệt phun, thì detector phải có khả năng chịu được áp suất cao (tới 6000 psi). Tuy nhiên detector UV có thể gần độc lập với HPLC của HPLC/TS/MS và trong trường hợp này áp suất HPLC tiêu chuẩn có thể được chấp nhận.
4.3 Thiết bị làm tinh khiết thuốc nhuộm azo chuẩn
4.3.1 Thiết bị chiết Soxhlet
4.3.2 Vòng chiết, độ dày đơn. 43 mm x 123 mm.
4.3.3 Giấy lọc, 9,0 cm (Whatman chất lượng loại 1 hoặc tương đương)
4.3.4 Cột silica-gel 3 in x 8 in nhồi silica gel (Loại 60. thuốc thử EM 70/230 mắt lưới).
4.4 Thiết bị chiết dùng cho hợp chất axit phenoxy clo hoá
4.4.1 Bình Erlenmeyer 500 mL miệng rộng Pyrex. 500 mL Pyrex, có khớp nối thuỷ tinh mài cỡ 24/40. 1000 mL Pyrex
4.4.2 Phễu tách 2000 mL
4.4.3 Ống đong có chia vạch 1000 mL
4.4.4 Phễu đường kính 75 mm
4.4.5 Máy lắc Wrist – Burrell Model 75 hoặc tương đương.
4.4.6 Máy đo pH.
4.5 Thiết bị cô mẫu Kuderna-Danish (K-D) (tùy chọn).
4.5.1 Ống cô 10 rnL- có chia vạch (Kontes K-570050-1025 hoặc tương đương). Dùng nút thuỷ tinh mài để tránh bay hơi dịch chiết.
4.5.2 Bình bay hơi 500 mL (Kontes K-570001-500 hoặc tương đương). Đi kèm theo bình cô là lo xo, kẹp hoặc tương đương.
4.5.3 Cột Snyder hai bi nhỏ (Kontes K-569001-0219 hoặc tương đương)
4.5.4 Lò xo 1/2 in (Kontes K-662750 hoặc tương đương)
CHÚ THÍCH Nên dùng các dụng cụ thủy tinh sau đây để thu hồi dung môi trong quá trình có yêu cầu sử dụng bình cô bay hơi Kuderna Danish. Qui định của địa phương hoặc nhà nước về sử dụng các thiết bị này giúp cho việc quản lý khí thoát ra của các chất hữu cơ dễ bay hơi. EPA khuyến nghị sử dụng các hệ thống tái sử dụng này như là biện pháp thực hiện chương trình giảm thải. Thu hồi dung môi là biện pháp để phù hợp với biện pháp giảm thiểu chất thải và ngăn chặn ô nhiễm bước đầu
4.5.5 Hệ thống thu hồi hơi dung môi (Kontes K-545000-1006 hoặc K-547300-0000, Thuỷ tinh Ace 6614-30, hoặc tương đương).
4.6 Pipet huyết thanh dùng một lần 5 mL x 1/10, 5,5 mm ID.
4.7 Ống thu gom 15 mL hình nón, có chia vạch (Kimble No. 45465 hoặc tương đương)
4.8 Lọ 5 mL hình nón, bằng thuỷ tinh có nút polytetrafluoroetylen (PTFE)-nút xoáy hoặc nắp trên ấn chặt.
4.9 Bông thuỷ tinh Supelco No. 2-0411 hoặc tương đương.
4.10 Bơm tiêm micro 100 mL, 50 mL, 10 mL (Hamilton 701 N hoặc tương đương) và 50 mL (Blunted, Hamilton 705SNR hoặc tương đương).
4.11 Máy bay hơi quay/máy cô quay có lắp thêm bình nhận 1000 ml.
4.12 Cân phân tích, 0,0001 g, mức tối đa là 0,01 g.
4.13 Bình định mức, Loại A 10 mL đến 1000 mL.
4.14 Ống đong chia vạch 100 mL.
4.15 Phễu tách 250 mL.
4.16 Phễu tách 2 lít, có nút PTFE.
4.17 Biến áp dùng cho máy cô (tuỳ chọn – dùng cho quá trình chiết cacbamat).
5. Thuốc thử
5.1 Hoá chất cấp độ tinh khiết hoá học cần được sử dụng trong tất cả phép thử. Ngoại trừ có những chỉ định khác, tất cả thuốc thử phải phù hợp vói các yêu cầu của Uỷ ban thuốc thử phân tích thuộc Hiệp hội hoá chất Hoa Kỳ. Các cấp độ khác có thể được sử dụng nếu chắc chắn các thuốc thử có đủ độ tinh khiết để cho phép sử dụng mà không làm giảm tính chính xác của phép phân tích.
5.2 Nước dùng cho thuốc thử không có chất hữu cơ. Tất cả những viện dẫn tới nước trong phương pháp này là nước thử không có chất hữu, như qui định trong Chương một.
5.3 Natri sunphat (hạt, khan), Na2SO4. Làm tinh khiết bằng cách sấy ở 400oC trong 4 giờ trong một khay nông, hoặc bằng cách làm sạch trước natri sunphat với metylen clorua.
5.4 Amoni axetat, NH4OOCCH3, dung dịch (0,1 M). Lọc qua màng lọc 0,45 micron (HA Millipore hoặc tương đương).
5.5 Axit axetic, CH3CO2H.
5.6 Dung dịch axit sunfuric.
5.6.1 (1:1. v/v) Rót từ từ 50 mL H2SO4 (sp. Gr. 1,84) vào 50 mL nước.
5.6.2 (1:3, v/v) Rót từ từ 25 mL H2SO4 (sp. Gr. 1,84) vào 75 mL nước.
5.7 Khí argon, tinh khiết 99 +%.
5.8 Dung môi
5.8.1 Metylen clorua. CH2CI2 – loại dùng cho thuốc bảo vệ thực vật hoặc tương đương
5.8.2 Toluen. C8H5CH3 – loại dùng cho thuốc bảo vệ thực vật hoặc tương đương.
5.8.3 Axeton, CH3COCH3 – loại dùng cho thuốc bảo vệ thực vật hoặc tương đương.
5.8.4 Dietyl Ete, C2H5OC2H5 – loại dùng cho thuốc bảo vệ thực vật hoặc tương đương. Phải chắc chắn không có peroxit bằng phép thử chiết (Định lượng EM hoặc tương đương). Qui trình để loại bỏ peroxit được thực hiện với phép thử chiết. Sau khi làm sạch, thêm vào mỗi lít ete 20 mL etyl alcohol bảo quản.
5.8.5 Metanol. CH3OH – loại dùng cho HPLC hoặc tương đương.
5.8.6 Axetonitril, CH3CN – loại dùng cho HPLC hoặc tương đương.
5.8.7 Etyl axetat CH3CO2C2H5 – loại dùng cho thuốc bảo vệ thực vật hoặc tương đương.
5.9 Vật liệu chuẩn – Vật liệu chuẩn tinh khiết hoặc dung dịch của từng chất phân tích đã được chứng nhận dùng cho phép phân tích. Thuốc nhuộm azo phải được làm tinh khiết trước khi sử dụng theo 5.10.
5.10 Làm sạch thuốc nhuộm azo
5.10.1 Quá trình này gồm hai bước: Đầu tiên, chiết Soxhlet thuốc nhuộm trong 24 giờ bằng toluen và sử dụng máy cô quay làm bay hơi dịch chiết lỏng đến khô hoàn toàn. Sau đó, chất rắn làm kết tinh lại từ toluen và được sấy khô trong lò ở khoảng 100 oC. Nếu bước này không đạt độ tinh khiết yêu cầu, cần thực hiện sắc ký cột. Cho chất rắn vào cột silica gel 3 x 8 in (4.3.4) và rửa giải bằng dietyl ete. Tách phần chưa tinh khiết qua sắc ký và thu lại phần thuốc nhuộm chính.
5.11 Dung dịch chuẩn gốc: Có thể chuẩn bị từ vật liệu chuẩn hoặc có thể mua dung dịch đã được chứng nhận. Dung dịch chuẩn gốc mua ngoài thị trường có thể được sử dụng nếu các dung dịch này được kiểm định đạt tiêu chuẩn EPA. Nếu các chuẩn EPA không có sẵn để kiểm chứng thì có thể sử dụng các dung dịch chuẩn đã được nhà sản xuất kiểm định và kiểm chứng với dung dịch chuẩn được chuẩn bị từ vật liệu tinh khiết.
5.11.1 Chuẩn bị dung dịch chuẩn gốc bằng cách cân chính xác 0,0100 g vật liệu tinh khiết. Hoà tan vật liệu này trong metanol hoặc dung môi phù hợp khác (ví dụ chuẩn bị Tris-BP trong etyl axetat), và pha loãng đến thể tích đã biết trong bình định mức.
CHÚ THÍCH Vi tính phản ứng của thuốc trừ cỏ clorin, các dung dịch chuẩn phải được chuẩn bị trong axetonitril. Sự metyl hoá sẽ xảy ra nếu chuẩn bị trong metanol.
Nếu độ tinh khiết của hợp chất được chứng nhận đạt 96 % hoặc cao hơn, có thể sử dụng lượng cân mà không cần hiệu chỉnh để tính toán nồng độ của dung dịch chuẩn gốc. Có thể sử dụng dung dịch chuẩn gốc mua ngoài thị trường ở bất kỳ nồng độ nào nếu các dung dịch này được nhà sản xuất hoặc bên thứ ba chứng nhận.
5.11.2 Chuyển dung dịch chuẩn gốc vào lọ thuỷ tinh có nút PTFE nút vặn hoặc nắp trên ấn chặt. Bảo quản ở 4 oC và tránh ánh sáng. Dung dịch chuẩn gốc cần phải kiểm tra thường xuyên các dấu hiệu phân huỷ hoặc bay hơi, đặc biệt trước khi chuẩn bị dung dịch hiệu chuẩn.
5.12 Dung dịch chuẩn hiệu chuẩn – cần được chuẩn bị bằng cách pha loãng dung dịch chuẩn gốc bằng metanol (hoặc dung môi phù hợp khác) với tối thiểu năm nồng độ khác nhau tương ứng với từng thông số quan tâm. Một trong những nồng độ này cần phải gắn với MDL, nhưng nằm trên MDL. Các nồng độ còn lại cần phải tương ứng với khoảng nồng độ dự kiến tìm được trong các mẫu thực hoặc cần phải định ra khoảng hoạt động của HPLC-UV/VIS hoặc HPLC-TS/MS. Các dung dịch chuẩn phải được thay thế sau một hoặc hai tháng, hoặc sớm hơn nếu thấy hỏng khi so sánh với dung dịch chuẩn kiểm tra.
5.13 Dung dịch chuẩn thay thế – Người phân tích cần phải giám sát thực hiện quá trình chiết, làm sạch (khi sử dụng), và hệ thống phân tích về hiệu quả của phương pháp đối với mỗi nền mẫu bằng cách thêm một hoặc 2 chuẩn thay thế vào từng mẫu, dung dịch chuẩn và dung dịch trắng (ví dụ hợp chất phospho hữu cơ hoăc hợp chất axit phenoxy clo hoá không được có trong mẫu).
5.14 Dung dịch chuẩn điều chỉnh HPLC/MS – Polyetylen glycol 400 (PEG-400), PEG-600, hoặc PEG- 800 nên được dùng làm dung dịch chuẩn điều chỉnh. Tuy nhiên, người phân tích có thể sử dụng dung dịch chuẩn điều chỉnh khác do nhà sản xuất thiết bị hoặc các nguồn đã được lập thành tài liệu khuyến nghị. Pha loãng đến 10 % (v/v) trong metanol nếu dùng dung dịch PEG. Việc sử dụng PEG phụ thuộc vào khoảng khối lượng phân tử chất phân tích: m.w. 500. thì dùng PEG-400; m.w. > 500, dùng PEG- 600 hoặc PEG-800.
5.15 Dung dịch nội chuẩn: Nếu dùng chuẩn nội chuẩn thì người phân tích nên sử dụng hợp chất đồng vị bền vững của cùng loại hoá chất nếu các chất này có sẵn (ví dụ 13C6-cacbofuran có thể được dùng làm dung dịch nội chuẩn trong phân tích cacbamat).
6. Lấy, bảo quản và xử lý mẫu
6.1 Xem nội dung giới thiệu của phần này, chất phân tích hữu cơ, 4.1.
7. Qui trình
7.1 Chuẩn bị mẫu: Mẫu để phân tích thuốc nhuộm azo phân tán và hợp chất phospho hữu cơ phải được chuẩn bị bằng Method 3500 thích hợp trước khi phân tích HPLC/MS.
Mẫu để phân tích Tris(2,3-dibromopropyl) phosphat nước thải phải được chuẩn bị theo 7.1.1 trước khi phân tích HPLC/MS. Mẫu để phân tích hợp chất clorin phenoxyaxit và các este của chúng cần phải được chuẩn bị theo 7.1.2 trước khi phân tích HPLC/MS.
7.1.1 Vi chiết cho Tris-BP
7.1.1.1 Mẫu rắn
7.1.1.1.1 Cân 1 g mẫu cho vào cốc mỏ. Nếu mẫu còn ẩm, thêm một lượng tương đương natri sunfat khan và trộn đều. Thêm 100 mL Tris-BP (nồng độ khoảng 1000 mg/L) vào mẫu đã chọn để thêm chuẩn. Thêm sao cho nồng độ cuối cùng phải là 100 ng/mL trong 1 mL dịch chiết.
7.1.1.1.2 Lấy bông thuỷ tinh ra khỏi pipet huyết thanh dùng một lần. Chèn 1 cm nút bông thuỷ tinh đã được silan hóa vào đáy (đầu hẹp) của pipét. Nhồi 2 cm natri sunfat khan lên trên bông thuỷ tinh Tráng rửa pipét và các thành phần trong pipét bằng 3 mL đến 5 mL metanol.
7.1.1.1.3 Cho mẫu vào pipet đã được chuẩn bị theo 7.1.1.1.2. Nếu vật liệu được nhồi trong pipét này bị khô thì trước tiên làm ẩm bằng vài mL metanol, sau đó cho mẫu vào pipét.
7.1.1.1.4 Chiết mẫu bằng 3 mL metanol đã chuẩn bị bằng 4 mL metanol/metylen clorua 50 % (v/v) (rửa cốc mẫu bằng từng lượng dung môi chiết trước khi thêm chúng vào pipet có chứa mẫu). Thu lấy dịch chiết vào ống thuỷ tinh chia vạch 15 mL.
7.1.1.1.5 Dùng kỹ thuật thổi dòng nitơ làm bay hơi dịch chiết đến 1 mL (7.1.1.1.6) Ghi lại thể tích này. Một số mẫu bùn có thể không làm bay hơi được đến nồng độ hợp lý.
7.1.1.1.6 Kỹ thuật thổi dòng nitơ
7.1.1.1.6.1 Đặt ống cô trong bếp cách thuỷ ấm (khoảng 35 oC) và dùng dòng nitơ khô, sạch, thổi nhẹ (đã lọc qua cột than hoạt tính) làm bay hơi thể tích dung môi đến mức yêu cầu.
CHÚ Ý Không dùng ống nhựa từ bẫy cacbon đến mẫu.
7.1.1.1.6.2 Thành trong của ống phải được rửa vài lần bằng metyl clorua trong khi thao tác. Trong quá trình bay hơi, mức dung môi trong ống phải được định vị để ngăn ngừa nước ngưng tụ vào mẫu (Ví dụ mức dung môi phải dưới mức nước bếp cách thuỷ). Không được để dịch chiết bị khô trong điều kiện vận hành bình thường. Tiến hành theo 7.1.1.1.7
7.1.1.1.7 Chuyển dịch chiết vào lọ thuỷ tinh có nút PTFE và bảo quản trong tủ lạnh ở 4 oC. Tiến hành phân tích với HPLC
7.1.1.1.8 Xác định phần trăm trọng lượng khô. Trong một vài trường hợp nhất định, kết quả của mẫu được yêu cầu dựa theo trọng lượng khô. Nếu yêu cầu các số liệu như vậy thì một phần mẫu dùng cho phép xác định này cần phải được cân cùng thời điểm khi cân phần dùng cho phép phân tích.
Cảnh báo Lò sấy cần phải đặt trong tủ hút hoặc được thông gió. Phòng thí nghiệm có thể bị nhiễm bẩn đáng kể khi sấy mẫu chất thải nguy hại bị nhiễm bẩn nặng.
7.1.1.1.9 Ngay sau khi cân mẫu để chiết, cân 5 g đến 10 g mẫu cho vào chén nung. Xác định phần trăm trọng lượng khô của mẫu bằng cách sấy qua đêm ở 105 oC. Để mát trong bình hút ẩm trước khi cân:
% trọng lượng khô = x 100
7.1.1.2 Mẫu nước
7.1.1.2.1 Sử dụng ống đong chia vạch 100 mL, lấy 100 mL mẫu và chuyển mẫu vào phễu tách 250 mL. Thêm 200 mL Tris-BP (nồng độ xấp xỉ 1000 mg/L) vào mẫu đã chọn để thêm chuẩn; lượng thêm vào cần phải có nồng độ cuối cùng là 200 ng/mL trong 1 mL dịch chiết.
7.1.1.2.2 Thêm 10 mL metylen clorua vào phễu tách. Đậy kín và lắc phễu tách ba lần, mỗi lần khoảng 30 giây, mở nắp sau mỗi lần lắc để giải phóng áp suất vượt quá.
CHÚ THÍCH Metylen clorua tạo áp suất cao rất nhanh; do vậy, cần phải thông hơi trước ngay sau khi phễu tách đã được gắn kín và lắc một lần. Metyl clorua là chất có khả năng gây ung thư, sử dụng các biện pháp phòng ngừa an toàn cần thiết.
7.1.1.2.3 Để lớp hữu cơ tách khỏi pha nước trong tối thiểu 10 phút. Nếu bề mặt phân tách nhũ tương giữa các lớp nhiều hơn một phần ba độ dày lớp dung môi, thì người phân tích phải tiến hành kỹ thuật cơ học để cho việc tách pha hoàn toàn. Xem 7.5, Method 3510.
7.1.1.2.4 Thu lấy dịch chiết vào ống thuỷ tinh có chia vạch 15 mL. Tiến hành như 7.1.1.1.5.
7.1.2 Chiết hợp chất clorin phenoxyaxit: Chuẩn bị mẫu đất, trầm tích và mẫu chất rắn khác phải theo Method 8151, trừ trường hợp không có sự thuỷ phân hoặc este hoá. (tuy nhiên, nếu người phân tích mong muốn xác định tất cả phenoxyaxit do axit, có thể thực hiện quá trình thuỷ phân). Điều 7.1.2.1 đưa ra sơ lược qui trình với các thay đổi cần thiết phù hợp để xác định bằng Method 8321. Điều 7.1.2.2 mô tả qui trình chiết đối với các mẫu nước.
7.1.2.1 Chiết các mẫu rắn
7.1.2.1.1 Cho 50 g mẫu rắn/trầm tích vào bình Erlenmeyer 500 mL miệng rộng. Thêm dung dịch đã thêm chuẩn nếu yêu cầu, lắc đều và để yên trong 15 phút. Thêm 50 mL nước thử không có chất hữu cơ và khuấy trong 30 phút. Xác định pH của mẫu bằng điện cực thuỷ tinh và pH met trong khi vẫn khuấy. Điều chỉnh pH đến 2 bằng H2SO4 lạnh (1:1), vừa khuấy, vừa kiểm tra pH trong 15 phút. Nếu cần, thêm H2SO4 cho đến khi pH đạt 2.
7.1.2.1.2 Thêm 20 mL axeton vào bình và lắc đều các thành phần bằng máy khuấy trong 20 phút. Thêm 80 mL dietyl ete vào cùng bình và lắc lại trong 20 phút. Gạn dịch chiết và đo thể tích dung môi thu hồi.
7.1.2.1.3 Dùng 20 mL axeton sau khi dùng 80 mL dietyl ete để chiết mẫu thêm hai lần. Sau khi thêm từng dung môi, hỗn hợp cần phải lắc bằng máy lắc trong 10 phút và gạn dịch chiết axeton-ete.
7.1.2.1.4 Sau khi chiết lần thứ ba, thể tích dịch chiết thu được cần phải ít nhất bằng 75 % thể tích dung môi thêm vào. Nếu không thực hiện được thì có thể cần phải chiết thêm. Cho dịch chiết vào phễu tách 2000 mL có chứa 250 mL natri sunfat 5 % đã được axit hoá. Nếu có dạng nhũ tương, thêm từ từ 5 g natri sunfat đã axit hoá (khan) cho đến khi hỗn hợp dung môi-nước tách hoàn toàn. Nếu cần. lượng natri sunfat đã được axit hoá được đưa vào tương đương với lượng mẫu.
7.1.2.1.5 Kiểm tra pH của dịch chiết. Nếu pH của dịch chiết £ 2, thì thêm HCI đậm đặc cho đến khi dịch chiết ổn định ở tại pH yêu cầu. Lắc nhẹ các thành phần trong phễu tách trong khoảng 1 phút và để cho các lớp tách hoàn toàn. Thu lấy pha nước vào trong bình mỏ sạch và chiết pha (lớp trên cùng) vào trong bình Erlenmeyer thuỷ tinh 500 mL. Rót pha nước vào lại phễu tách và chiết lại dùng 25 mL dietyl ete. Để cho các lớp tách hoàn toàn và loại bỏ lớp nước. Gộp dịch chiết ete vào bình Erlenmeyer 500 mL ở trên
7.1.2.1.6 Thêm 45 g đến 50 g natri sunfat khan đã được axit hoá vào hỗn hợp dịch chiết ete. Để dịch chiết tiếp xúc với natri sunfat trong khoảng 2 giờ.
CHÚ THÍCH Bước làm khô có tính quyết dịnh. Hơi ẩm còn lại trong ete sẽ dẫn đến độ thu hồi thấp. Lượng natri sunfat được dùng là vừa đủ nếu nhìn thấy một số tinh thể tự do dưới đáy khi khuấy bình. Nếu tất cả natri sunfat đóng rắn thành một bánh, thì thêm vài gam natri sunfat đã axit hoá và kiểm tra lại bằng cách khuấy đều. Thời gian làm khô 2 giờ là tối thiểu, tuy nhiên, dịch chiết có thể để qua đêm trong khi làm khô bằng natri sunfat
7.1.2.1.7 Chuyển dịch chiết ete bằng phễu lọc có nhồi bông thuỷ tinh đã rửa axit vào bình K-D 500 mL có lắp ống ngưng 10 mL. Dùng que thuỷ tinh để khuấy hoà natri sunfat trong khi chuyển. Rửa bình Erlenmeyer và cột bằng 20 mL đến 30 mL dietyl ete để chuyển định lượng hoàn toàn. Giảm thể tích dịch chiết dùng kỹ thuật vi K-D (xem 7.1.2.1.8).
7.1.2.1.8 Thêm một hoặc hai mảnh sỏi sạch vào bình và gắn vào cột Snyder ba bóng. Gắn bình thuỷ tinh thu hồi hơi dung môi (thiết bị ngưng và thu hồi) (xem 4.5.5) vào cột Snyder của thiết bị K-D theo hướng dẫn của nhà sản xuất. Làm ướt cột Snyder trước bằng cách thêm khoảng 1 mL dietyl ete từ đỉnh xuống. Đặt thiết bị trong bếp cách thuỷ nóng (60 oC đến 65 oC) sao cho một phần của ống cô được nhúng trong nước nóng và toàn bộ mặt dưới bình được tiếp xúc với hơi nước. Điều chỉnh nhiệt độ nước và vị trí của thiết bị theo chiều thẳng đứng như yêu cầu cho đến khi quá trình cô hoàn thành trong 15 phút đến 20 phút. Tại tốc độ chưng cất phù hợp, bóng của cột sẽ phát tiếng, nhưng buồng sẽ không bị chảy tràn. Khi thể tích chất lỏng đạt 5 mL, lấy thiết bị K-D khỏi bếp cách thuỷ và để cho ráo nước và để nguội trong ít nhất 10 phút.
7.1.2.1.9 Thay dung môi của dịch chiết bằng axetonitril bằng cách chuyển định lượng dịch chiết dùng axetonitril vào thiết bị phun xuống. Thêm 5 mL axetonitril. Giảm thể tích dịch chiết theo 7.1.1.1.6 và điều chỉnh thể tích cuối cùng xuống 1 mL.
7.1.2.2. Chuẩn bị mẫu nước
7.1.2.2.1 Dùng bình hình trụ có chia vạch 1000 mL, đong 1 lít mẫu, ghi thể tích mẫu chính xác đến 5 mL và chuyển chúng vào phễu tách. Nếu dự đoán nồng độ cao, có thể sử dụng thể tích nhỏ hơn và sau đó pha loãng bằng nước không có chất hữu cơ đến 1 lít. Điều chỉnh pH đến nhỏ hơn 2 bằng axit sunfuric (1:1).
7.1.2.2.2 Thêm 150 mL dietyl ete vào chai mẫu, đậy kín và lắc trong 30 giây để rửa thành lọ. Chuyển dung môi rửa vào phễu tách và chiết mẫu bằng cách lắc phễu trong 2 phút có thông hơi để giảm áp suất vượt quá. Để yên cho lớp hữu cơ tách khỏi lớp nước trong khoảng thời gian tối thiểu 10 phút. Nếu bề mặt phân tách nhũ tương giữa các lớp nhiều hơn một phần ba độ dày lớp dung môi, thì người phân tích phải tiến hành kỹ thuật cơ học để tách pha hoàn toàn. Kỹ thuật tối ưu phụ thuộc vào mẫu và có thể bao gồm cả kỹ thuật khuấy, loc nhũ tương qua bông thuỷ tinh, ly tâm hoặc các phương pháp vật lý khác. Tháo pha nước vào bình thót cổ Erlenmeyer 1000 mL.
7.1.2.2.3 Lặp lại qui trình chiết thêm hai lần, mỗi lần dùng 100 mL dietyl ete. Gộp tất cả các dịch chiết vào bình Erlenmeyer 500 mL. (Rửa bình 1000 mL bằng từng lượng dung môi chiết để tạo thành dung dịch chuyển định lượng).
7.1.2.2.4 Tiến hành theo 7.1.2.1.6 (làm khô, cô mẫu K-D, thay đổi dung môi và điều chỉnh thể tích cuối cùng).
7.1.3 Chiết cacbamat: Chuẩn bị mẫu đất, trầm tích và mẫu chất rắn khác phải theo Method 3500 phù hợp
7.1.3.1 Sử dụng Method 3500 phù hợp để chiết 40 g mẫu bằng metyl clorua.
7.1.3.2 Thực hiện các bước cô đặc bằng cách sử dụng máy cô quay hoặc K-D, đến thể tích 5-10 mL.
7.1.3.3 Nên sử dụng bộ chuyển đổi trên máy cô quay để đạt nồng độ cuối cùng và dung môi thay đổi đến 1 mL thể tích cuối cùng metanol. Nếu bộ chuyển đổi không có sẵn, có thể sử dụng dòng nitơ thổi nhẹ trong tủ hút để đạt được nồng đô cuối cùng này.
7.1.4 Chiết cacbamat: Chuẩn bị mẫu nước phải theo Method 3500 phù hợp.
7.1.4.1 Dùng Method 3500 phù hợp để chiết một lít mẫu bằng metyl clorua.
7.1.4.2 Cô mẫu và đổi sang metanol giống như áp dụng theo 7.1.3.2 và 7.1.3.3.
7.2 Trước khi phân tích HPLC, dung môi chiết phải được thay bằng metanol hoặc axetonitril (7.1.2.1.9). Việc thay đổi được thực hiện bằng việc sử dụng qui trình K-D đã nêu trong tất cả các phương pháp chiết.
7.3 Điều kiện sắc ký HPLC
7.3.1 Điều kiện sắc ký phân tích các chất đặc thù được trình bày trong Bảng 1. Các điều kiện sắc ký không phải là phân tích các chất đặc trưng như sau
Tốc độ dòng 0.4 mL/min
Pha động sau cột: 0,1 M amoni axetat (1% metanol)
(0,1 M amoni axetat đối với hợp chất phenoxyaxit)
Tốc độ dòng sau cột: 0,8 mL/min
7.3.2 Nếu có vấn đề sắc ký từ hợp chất còn giữ lại khi phân tích đối với thuốc nhuộm azo, hợp chất phospho hữu cơ và tris(2,3-dibromopropyl)phosphat, có thể áp dụng dòng ổn định metylen clorua 2 % nếu cần. Dung dịch metylen clorua/metanol phải được dùng cẩn thận như là dung dịch rửa giải HPLC. Axit axetic (1 %), pha động loại khác có thể được dùng với hợp chất có nhóm chức axit.
7.3.3 Tốc độ dòng tổng số 1,0 mL/min đến 1,5 mL/min là cần thiết để duy trì sự ion hoá phun nhiệt.
7.3.4 Thời gian lưu đối với hợp chất phospho hữu cơ trên cột phân tích đặc trưng được trình bày trong Bảng 9.
7.4 Các điều kiện vận hành HPLC/Phun nhiệt/MS nên áp dụng: Trước khi phân tích mẫu, người phân tích phải đánh giá độ nhạy tương đối của các hợp chất cần phân tích đối với chế độ ion hoá để tìm ra chế độ cho độ nhạy tốt hơn trong quá trình phân tích. Việc đánh giá này dựa trên cấu trúc của chất phân tích hoặc bằng các phân tích đang tiến hành trong từng cặp chế độ ion hoá. Xem thêm 7.5.2.6 về vấn đề này.
7.4.1 Chế độ ion hoá dương
Chất xua đuổi (dây hoặc đĩa, tuỳ chọn): 170 V đến 250 V (độ nhạy đã được tối ưu hoá) Xem Hình 2 về sơ đồ của nguồn có chất xua đuổi dây.
Điện cực phóng: tắt
Dây bật hoặc tắt (tuỳ chọn, phụ thuộc chất phân tích)
Khoảng khối lượng: 150 amu đến 450 amu (phụ thuộc chất phân tích, dự đoán cao hơn khối lượng phân tử của hợp chất từ 1 amu đến 18 amu)
Thời gian quét: 1,50 giây/1 lần quét
7.4.2 Chế độ ion hoá âm
Điện cực phóng: bật
Dây: tắt
Khoảng khối lượng: 135 amu đến 450 amu
Thời gian quét: 1,50 giây/1 lần quét
7.4.3 Nhiệt độ phun nhiệt
Bộ kiểm soát hơi: 110 oC đến 130 oC
Đầu phun hơi: 200 oC đến 215 oC.
Dòng. 210 oC đến 220 oC.
Khối nguồn: 230 oC đến 265 oC (Một số hợp chất có thể phân huỷ trong khối nguồn ở nhiệt độ cao, người vận hành cần có kiến thức về đặc tính hoá học để đánh giá nhiệt độ nguồn phù hợp).
7.4.4 Thể tích mẫu bơm: Thường sử dụng thể tích 20 đến 100 mL . Nếu thực hiện bằng tay, vòng bơm phải được bơm đầy tối thiểu là hệ số của 2 (ví dụ 20 ml mẫu được dùng để bơm đầy vòng bơm 10 ml). Nếu chất rắn có trong dịch chiết, để các chất rắn này lắng hoặc ly tâm dịch chiết và lấy thể tích để bơm từ lớp sạch.
7.5 Hiệu chuẩn
7.5.1 Hệ thống phun nhiệt/MS: Phải là phần mềm dò trên bốn cực 1 (và bốn cực 3 đối với quét bốn cực 3 lần) để cho chính xác khối lượng, độ nhạy và độ phân giải. Nên tiến hành dùng polyetylen glycol (PEG) 400, 600 hoặc 800 (xem 5.14) có khối lượng phân tử trung bình tương ứng là 400, 600 và 800. Người phân tích có thể dùng dung dịch chuẩn khác theo khuyến nghị của nhà sản xuất thiết bị hoặc các nguồn tài liệu khác. Nếu dùng PEG, hỗn hợp các PEG có thể được tạo thành sao cho hỗn hợp có khối lượng xấp xỉ bằng khối lượng cần có sử dụng trong các phân tích. Dùng PEG 400 cho phân tích hợp chất clorin phenoxyaxit PEG được đưa vào qua bề mặt phun nhiệt, phá vỡ HPLC.
7.5.1.1 Các thông số hiệu chuẩn khối lượng như sau:
Đối với PEG 400 và 600 . Đối với PEG 800
Khoảng khối lượng: 15 amu đến 765 amu Khoảng khối Iượng: 15 amu đến 900 amu
Thời gian quét: 0,5 giây đến 5,0 giây/1 lần quét Thời gian quét: 0,5 giây đến 5,0 giây/1 lần quét
Với 2 đến 3 lần bơm cần phải thực hiện khoảng 100 lần quét. Kết quả quét phù hợp nhất với bảng khối lượng chính xác (xem Bảng 7 và 8) phải được dùng như là bảng hiệu chuẩn. Nếu sử dụng chất hiệu chuẩn không phải PEG, khoảng khối lượng cần phải cao hơn khối lượng cao nhất đã dùng để hiệu chuẩn từ 15 amu đến khoảng 20 amu. Thời gian quét cần được chọn sao cho sẽ có ít nhất 6 lần quét dọc theo pic hiệu chuẩn.
7.5.1.2 Khoảng khối lượng thấp từ 15 amu đến 100 amu bao gồm các ion trong dung dịch đệm amonium axetat được dùng trong quá trình phun nhiệt: NH4+ (18 amu), NH4+H2O (36), CH3OH NH4+ (50) (metanol), hoặc CH3CN NH4+ (59) (axetonitril) và CH3OOH NH4+ (78) (axit axetic). Sự xuất hiện số khối ion m/z 50 hoặc 59 phụ thuộc vào việc sử dụng metanol hay axetonitril làm chất hữu cơ bổ sung. Khoảng khối lượng cao hơn sẽ bao gồm các ion amoni gắn vào các etylen glycol (ví dụ H(OCH2CH2)nOH nếu n =4, cho ion H(OCH2CH2)4OH NH4‘ tại số khối m/z 212).
7.5.2 Sắc ký lỏng
7.5.2.1 Chuẩn bị dung dịch chuẩn như trong 5.12
7.5.2.2 Lựa chọn các điều kiện ion hoá phù hợp như trong 7 4. Bơm từng dung dịch chuẩn vào HPLC, sử dụng các điều kiện sắc ký như trong Bảng 1. Tham khảo điều 7 của Method 8000 về hướng dẫn lựa chọn nội chuẩn và ngoại chuẩn và các tiêu chí hiệu chuẩn cho phép. Hệ số tương quan điều chỉnh (r2) ít nhất 0,97 cần được sử dụng cho chất phân tích clorin phenoxyaxit. Trong phần lớn trường hợp các ion kép (M’H)’ và (M’NH4)’ là những ion chiếm đa số. Ví dụ, Bảng 9 liệt kê thời gian lưu và các ion chính (> 5%) có trong phổ tứ cực đơn nhiệt phun ion hoá dương của các hợp chất phospho hữu cơ.
7.5.2.2.1 Sử dụng giám sát ion có chọn lọc (SIM) được chấp nhận trong các trường hợp yêu cầu giới hạn phát hiện nằm dưới khoảng thông thường của phân tích phổ đầy đủ. Tuy nhiên, SIM có thể cho độ tin cậy thấp hơn trong nhận dạng hợp chất trừ khi các đa ion được giám sát/monitoring đối với từng hợp chất.
7.5.2.2.2 Việc sử dụng monitoring phản ứng có chọn lọc (SRM) cũng được chấp nhận khi dùng MS/MS 3/4 và cần phải nâng cao độ nhạy.
7.5.2.3 Nếu sử dụng HPLC-UV, phải hiệu chuẩn thiết bị bằng cách chuẩn bị dung dịch chuẩn theo 5.12 và bơm từng dung dịch chuẩn vào HPLC dùng các điều kiện sắc ký như trong Bảng 1. Hợp nhất tất cả các vùng có sắc ký cao nhất cho từng nồng độ. Định lượng bằng HPLC-UV có thể được thực hiện nếu đã biết cản trở mẫu và/hoặc chất phân tích cùng rửa giải không có ảnh hưởng.
7.5.2.4 Đối với các phương pháp đã qui định trong 7.5.2.2 và 7.5.2.3, thời gian lưu pic sắc ký là một biến số quan trọng trong nhận dạng chất phân tích. Do vậy, tỉ số giữa thời gian lưu của mẫu phân tích với chất chuẩn cần phải bằng 1,0 đến 0,1.
7.5.2.5 Nồng độ của mẫu phân tích sẽ được xác định bằng cách dùng đường chuẩn theo 7.5.2.2 và 7.5.2.3. Các đường chuẩn này phải được dựng trong cùng ngày với từng mẫu được phân tích. Các mẫu có nồng độ vượt quá khoảng chuẩn cần phải được pha loãng để nồng độ nằm trong khoảng này.
7.5.2.6 Khi sử dụng MS hoặc MS/MS, và nếu phù hợp với các chất cần phân tích và mục tiêu phân tích, việc xác định cả ion hoá ion dương và ion âm có thể được thực hiện trên từng dịch chiết mẫu. Tuy nhiên, một số nhóm hợp chất quan tâm sẽ cho độ nhạy tốt hơn nhiều khi chỉ ion hoá ion dương hoặc ion âm, dẫn đến phân tích đơn (ví dụ cacbamat nhạy hơn với chế độ ion hoá ion dương và phenoxyaxit thì nhạy hơn với chế độ ion hoá ion âm). Trước khi phân tích mẫu, người phân tích cần đánh giá độ nhạy tương đối của các hợp chất cần phân tích với từng chế độ ion hoá để xác định chế độ nào có thể cho độ nhạy tốt hơn trong phân tích. Việc đánh giá này có thể dựa trên cấu trúc chất phân tích hoặc bằng tiến hành phân tích trong từng cặp chế độ ion hoá.
7.5.2.7 Tham khảo Method 8000 về thông tin tính toán nồng độ mẫu và các thông số QC như độ chuẩn và độ chụm.
7.5.2.8 Độ chuẩn cũng có thể được tính từ tỉ số cho tín hiệu (diện tích) với lượng bơm vào. Tỉ số này được định nghĩa là hệ số hiệu chuẩn (CF) đối với từng nồng độ chuẩn. Nếu độ lệch chuẩn tương đối theo phần trăm (% RSD) của CF nhỏ hơn 20 % của khoảng làm việc, có thể giả định độ tuyến tính qua gốc toạ độ, và hệ số hiệu chuẩn trung bình có thể được dùng trong khoảng của đường chuẩn. CF và % SRD có thể được tính như sau:
CF = Tổng diện tích của pic/ khối lượng bơm vào (ng)
![]()
Trong đó
SD là độ lệch chuẩn các giữa CF
![]() là giá trị CF trung bình
là giá trị CF trung bình
7.6 Phân tích mẫu
7.6.1 Việc phân tích mẫu được thực hiện sau khi hệ thống LC/MS được hiệu chuẩn như các bước trong 7.5. Mẫu trước tiên nên được phân tích trong chế độ ion hoá ion âm. Nếu dự đoán mức hợp chất thấp thì mẫu cần phải quét trong chế độ ion hoá ion dương.
7.6.1.1 Bơm mẫu trắng (metanol) cần được phân tích sau khi phân tích dung dịch chuẩn, để xác định bất cứ sự nhiễm bẩn nào còn lại trong hệ thống Phun nhiệt/HPLC/MS.
7.6.1.2 Nếu thực hiện bơm mẫu thủ công, lấy phần mẫu phù hợp như 7.4.4. Khởi dộng rửa giải gradien HPLC, tải và bơm phần mẫu và bắt đầu phân tích hệ thống dữ liệu phổ khối lượng.
7.6.1.3 Nếu dùng bơm tự động, đảm bảo rằng thiết bị được cài đặt phù hợp với hướng dẫn của nhà sản xuất và tất cả các mẫu và dung dịch chuẩn được đưa vào theo thứ tự phù hợp. Khởi động bộ bơm mẫu tự động, rửa giải gradien HPLC và hệ thống dữ liệu ghi phổ.
7.7 Tính toán
7.7.1 Dùng qui trình hiệu chuẩn nội chuẩn hoặc ngoại chuẩn (Method 8000). xác định nhận dạng và định lượng từng pic thành phần trong sắc đồ ion của mẫu tương ứng với hợp chất được dùng cho qui trình hiệu chuẩn. Xem Method 8000 về còng thức tính toán.
7.7.2 Thời gian lưu của pic sắc ký là thông số quan trọng đối với nhận dạng chất phân tích. Tuy nhiên do nền mẫu có thể thay đổi điều kiện cột sắc ký nên thời gian lưu khônq có ý nghĩa, và việc xác nhận phổ khối lượng là tiêu chí quan trọng trong nhận dang chất phân tích.
7.8 Phân tích xác nhận phun nhiệt HPLC/MS/MS tùy chọn
7.8.1 Tương ứng với phương pháp này, MS/MS cần phải được coi là va chạm ion con đã được hoạt tính phân ly từ tứ cực một trên một khối lượng (ion mẹ), từ cực hai áp lực với argon và với điện thế lớn hơn bình thường, tử cực ba quét trên phạm vi kỳ vọng
7.8.2 Vì qui trình phun nhiệt chỉ phóng ra một hoặc hai ion trên một hợp chất nên sử dụng MS/MS có chệ độ vận hành cụ thể hơn thu được thông tin cấu trúc phân tử. Trong chế độ này, quét nhanh mẫu có thể thực hiện thông qua việc bơm trực tiếp mẫu vào phun nhiệt
7.8.3 Đối với thí nghiệm MS/MS, tứ cực thứ nhất cần phải được đặt để hút các phân tử proton hoặc các ammoni của các chất phân tích quan tâm. Tứ cực thứ ba cần phải quét từ 30 amu đến khoảng trên của các phân tử proton.
7.8.4 Áp suất khí va chạm (Ar) cần được đặt khoảng 1,0 m Torr và năng lượng va chạm ở 20 eV. Nếu các thông số này không đủ để tạo ra đáng kể sự phá vỡ ra từng mảnh, thì có thể tăng giá trị thông số này lên trên giá trị đã đặt để tạo ra sự va chạm nhiều và mạnh hơn nữa.
7.8.5 Đối với các phép phân tích xác định, pic cơ bản của phổ va chạm cần phải được xác định như là ion định lượng. Đối với các phân tích đặc biệt hơn, nên chọn một ion thứ hai làm ion định lượng bổ sung.
7.8.6 Dựng đường hiệu chuẩn như trình bày trong 7.5.2.
7.8.7 Sự nhiễm bẩn và cản trở MS/MS
7.8.7.1 Nếu chế độ MS/MS được dùng không có sự phân tách sắc ký (quét nhanh), phương pháp phân tích mẫu trắng phải chứng tỏ việc chuẩn bị mẫu và qui trình phân tích là không bị nhiễm bẩn bởi các chất cần phân tích hoặc các hợp chất cản trở. Tham khảo điều 8 của Method 8000 về hướng dẫn qui trình phương pháp mẫu trắng được chấp nhận. Nếu sự nhiễm bẩn được phát hiện trong phương pháp mẫu trắng vượt giới hạn cho phép, thì cần thiết phải chuẩn bị lại và phân tích lại các mẫu bị ảnh hưởng
7.8.7.2 Phổ MS/MS của dung dịch chuẩn và mẫu có thể được so sánh và kiểm tra tỉ số của ba ion chính (nồng độ lớn nhất). Các tỉ số này cần phải xấp xỉ như nhau trừ khi có cản trở. Nếu xuất hiện cản trở, sắc ký phải được sử dụng.
7.8.7.3 Tín hiệu của chất cần phân tích trong mẫu có thể bị kìm hãm bởi các chất cản trở cùng chiết mà các ion được quan trắc sẽ không cho tín hiệu. Để giám sát sự kìm hãm tín hiệu này, có thể thêm dung dịch nội chuẩn vào tất cả các dung dịch chuẩn, mẫu trắng và dịch chiết mẫu ở nồng độ phù hợp trước khi phân tích. Dung dịch nội chuẩn có thể là bất kỳ hợp chất nào có phản ứng tốt trong chế độ ion hoá tương ứng và không tìm thấy trong tự nhiên. (Chú thích: d5-atrazin được dùng rất thành công khi phân tích ion dương, còn d3-2,6-dinitrotoluen lại rất thành công khi phân tích ion âm). Lượng thêm vào cần phải chọn sao cho tín hiệu sinh ra ít nhất bằng 100 lần mức nhiễu đối với từng ion. Tín hiệu của dung dich nội chuẩn cần phải được giám sát. Yêu cầu phải phân tích lại với bất kỳ mẫu nào mà chiều cao pic dung dịch nội chuẩn thay đổi nhiều hơn 30 % so với chiều cao trung bình nội chuẩn thu được trong quá trình hiệu chuẩn năm điểm. Nếu kết quả phân tích lại khẳng định sự thay đổi này trong tín hiệu, phải phân tích lại mẫu bằng tách sắc ký. Có thể sử dụng nội chuẩn này để định lượng nồng độ chất phân tích. Việc định lượng ngoại chuẩn cũng được cho phép.
7.8.8 Với các nồng độ chưa biết, tổng diện tích của (các) ion được tính và sử dụng đường chuẩn đưa ra như trong 7.5 để thu được số lượng bơm.
7.8.9 Kỹ thuật MS/MS cũng có thể được dùng để thực hiện phân tích cấu trúc trên các ion đại diện bởi tỉ số m/z không xác định được. Qui trình đối với hợp chất chưa biết cấu trúc là thiết lập một thí nghiệm CAD lên ion quan tâm. Phổ được hình thành từ thí nghiệm này biểu thị cấu trúc của hợp chất qua sự vỡ ra từng mảnh của nó. Những người có chuyên môn về phổ khối lượng và một số thông tin về lịch sử của mẫu là cần thiết khi diễn giải phổ. (thí nghiệm CAD về tiêu chuẩn thực sự về các hợp chất dự đoán là cần thiết để khẳng định hoặc phủ nhận các chất này).
7.9 Xác nhận CAD dùng dây xua đuổi tùy chọn.
7.9.1 Xem Hình 3 về vị trí chính xác dây xua đuổi dạng vòng trong nguồn phun nhiệt.
7.9.2 Khi dây xua đuổi dạng vòng được lắp vào dòng phun nhiệt, điện thế có thể tăng tới xấp xỉ 500 V đến 700 V. Cần phải có điện thế đủ lớn để tạo ra các phân đoạn ion nhưng cũng không được quá lớn làm rút ngắn quá trình
7.9.3 Tiếp tục theo các bước trong 7.6.
8. Kiểm soát chất lượng
8.1 Tham khảo Chương một và Method 8000 về qui trình kiểm soát chất lượng cụ thể (QC). Mỗi phòng thí nghiệm cần phải duy trì chương trình đảm bảo chất lượng chính thức. Phòng thí nghiệm cũng cần lưu giữ các kết quả để lập thành tài liệu chất lượng các dữ liệu.
8.2 Qui trình kiểm suất chất lượng cần cho đánh giá vận hành hệ thống HPLC được trình bày trong Method 8000 phần 7 và bao gồm cả đánh giá thời gian lưu, kiểm định hiệu chuẩn và phân tích sắc ký của mẫu. Kiểm tra qui trình thực hiện của toàn bộ hệ thống phân tích hằng ngày sử dụng dữ liệu thu được từ các phân tích trắng, chuẩn và mẫu nhắc lại.
8.2.1 Xem 7.5.2.8 về các thông số HPLC/MS đối với đường chuẩn giới hạn RSD%.
8.2.2 Xem 7.5.2.4 về giới hạn QC thời gian lưu
8.2.3 Nếu không đáp ứng được bất kỳ giới hạn QC sắc ký nào, người phân tích cần phải kiểm tra hệ thống LC đối với:
– Rò rỉ
– Phân phối áp suất phù hợp,
– Cột bảo vệ bị bẩn, có thể cần thay thế hoặc nhồi lại, và
– Sự bít phun nhiệt từng phần có thể xảy ra.
Nếu một trong các trường hợp kể trên xảy ra cần phải ngừng hệ thống HPLC/TS, sửa chữa và hoặc thay thế, và sau đó bắt đầu lại phân tích. Dung dịch chuẩn hiệu chuẩn cần phải được phân tích lại trước khi phân tích mẫu, như mô tả trong 7.5.
8.2.4 Kinh nghiệm của người phân tích thực hiện sắc kỷ lỏng là vô cùng quý giá cho sự thành công của phương pháp này cần phải đánh giá hiệu chuẩn hằng ngày để xác định hệ thống sắc ký được vận hành phù hợp cho mỗi ngày thực hiện phân tích. Nếu hệ thống có bất kỳ thay đổi nào (ví dụ thay đổi cột), thì hệ thống phải được hiệu chuẩn lại.
8.3 Chứng minh ban đầu về tính hiệu quả. Mỗi phòng thí nghiệm phải chứng tỏ hiệu quả ban đầu với từng việc chuẩn bị mẫu và phương pháp kết hợp xác định để dữ liệu thu được từ các chất cần phân tích trong một nền mẫu sạch có độ chuẩn và độ chụm cho phép. Phòng thí nghiệm cũng phải lặp lại các thao tác tương tự khi có các nhân viên mới được đào tạo hoặc có những thay đổi đáng kể trong các thiết bụ đo. Xem Phương pháp 8000 phần 8.0 về thông tin hướng dẫn việc chứng minh.
8.4 Kiểm soát chất lượng mẫu để chuẩn bị và phân tích: phòng thí nghiệm cũng phải có thủ tục để lập thành tài liệu các ảnh hưởng của nền mẫu lên phương pháp thực hiện (độ chụm, độ chính xác và giới hạn phát hiện). Tối thiểu phải bao gồm phân tích QC các mẫu kể cả mẫu trắng, mẫu đã thêm chuẩn, mẫu đúp và mẫu kiểm soát phòng thí nghiệm (LCS) trong từng mẻ phân tích và thêm chuẩn thay thế vào từng mẫu hiện trường và mẫu QC.
8.4.1 Thành lập tài liệu về các ảnh hưởng của nền mẫu cần phải bao gồm ít nhất các phân tích: một mẫu đã thêm chuẩn và một mẫu kép chưa thêm chuẩn hoặc một mẫu nên thêm chuẩn/mẫu kép thêm chuẩn. Quyết định chuẩn bị và phân tích mẫu kép hay mẫu đã thêm chuẩn/ mẫu kép đã thêm chuẩn phải được dựa trên những thông tin về mẫu trong loạt mẫu. Nếu mẫu được dự đoán có chứa các chất cần phân tích, thì phòng thí nghiệm có thể dùng một mẫu nền thêm chuẩn và phân tích mẫu kép của một mẫu hiện trường chưa thêm chuẩn. Nếu mẫu dự đoán không chứa chất cần phân tích thì phòng thí nghiệm cần phải sử dụng một mẫu nền thêm chuẩn và cặp mẫu kép thêm chuẩn.
8.4.2 Mẫu kiểm soát phòng thí nghiệm (LCS) cần phải có trong từng loạt phân tích LCS có chứa phần mẫu thử của nền mẫu sạch (đối chứng) tương tự với nền mẫu và có cùng trọng lượng và thể tích LCS được thêm chuẩn với cùng chất phân tích tại cùng nồng độ với nền thêm chuẩn. Nếu kết quả của nền mẫu thêm chuẩn cho thấy có thể có vấn đề chính tại nền mẫu thêm chuẩn thì kết quả LCS được dùng để kiểm định phòng thí nghiệm có thể thực hiện phân tích trong một nền mẫu sạch
8.4.3 Xem Method 8000. điều 8 về chi tiết tiến hành thủ tục kiểm soát chất lượng mẫu trong chuẩn bị và phân tích
8.5 Độ thu hồi chất thay thế: Phòng thí nghiệm phải đánh giá dữ liệu độ thu hồi chất thay thế từ từng mẫu so với giới hạn kiểm soát chất thay thế được xây dựng bởi phòng thí nghiệm. Xem Method 8000, điều 8 để có thông tin đánh giá dữ liệu thay thế và xây dựng, cập nhật các giới hạn thay thế.
Phòng thí nghiệm nên có các bước thực hiện đảm bảo chất lượng bổ sung đôi khi sử dụng phương pháp này. Các bước thực hành cụ thể phụ thuộc vào nhu cầu của phòng thí nghiệm và bản chất của mẫu. Nếu cần, phòng thí nghiệm cần phải phân tích vật liệu chuẩn tham khảo và tham gia vào các nghiên cứu đánh giá đặc tính tương ứng.
9. Phương pháp tiến hành
9.1 Các nghiên cứu độ chính xác và độ chụm của người thao tác đã được tiến hành với mẫu trầm tích thêm chuẩn, mẫu nước thải, mẫu bùn và mẫu nước. Kết quả được trình bày ở Bảng 4, 5, 6, 11, 12, 15, 20 và 21. Bảng 4, 5 và 6 đưa ra các dữ liệu của phòng thí nghiệm cho Red disperse 1, Bảng 11 đưa ra dữ liệu cho hoá chất bảo vệ thực vật phospho hữu cơ, Bảng 12 cho Clo tris-BP, Bảng 15 cho thuốc trừ cỏ axit clorophenoxy và Bảng 20, 21 cho cacbamat.
9.2 LOD cần phải tính đối với các chất phân tích đã biết, trên từng thiết bị được sử dụng. Bảng 3, 10 và 13 liệt kê giới hạn phát hiện (LOD) và/hoặc giới hạn định lượng ước lượng (EQL) điển hình với phương pháp này.
9.2.1 LOD trình bày trong phương pháp này được tính dựa trên phân tích lặp lại 3 lần với bốn nồng độ chuẩn với nồng độ thấp nhất nằm gần giới hạn phát hiện của thiết bị. Phép hồi qui tuyến tính được thực hiện trên dữ liệu tính toán độ dốc và điểm cắt. Ba lần độ lệch chuẩn (3s) của lượng chuẩn thấp nhất theo độ dốc và điểm cắt tính toán được sử dụng để tìm LOD. LOD không được tính toán theo các chi tiết trong Chương một, nhưng theo hướng dẫn ACS qui định trong Tài liệu tham khảo 4.
9.2.2 Bảng 17 trình bày so sánh các LOD theo Method 8151 và Method 8321 đối với hợp chất axit phenoxy clo hoá
9.3 Bảng 16 trình bày số liệu độ chuẩn và độ chụm của phép thử liên phòng thí nghiêm đối với thuốc trừ cỏ axit clorophenoxy. Dữ liệu tổng hợp này được dựa trên số liệu từ ba phòng thí nghiệm phân tích dung dịch dung môi lặp lại hai lần tại từng nồng độ được quy định trong bảng.
9.4 Bảng 22 và 23 trình bày số liệu độ chuẩn và độ chụm của phép thử liên phòng thí nghiệm đối với cacbamat. Dữ liệu tổng hợp này được dựa trên số liệu từ chín phòng thí nghiệm đã phân tích các dung dịch dung môi lặp lại ba lần tại từng nồng độ được quy định trong các bảng.
Bảng 1 – Điều kiện sắc ký HPLC nên dùng
|
Pha động ban đầu (%) |
Thời gian ban đầu (min) |
Gradien cuối cùng (tuyến tính) (min) |
Pha động cuối cùng (%) |
Thời gian (min) |
|
Chất phân tích: |
|
ỉ |
||
|
Hợp chất phospho hữu cơ |
|
|
|
|
|
50/50 (nước/metanol) |
0 |
10 |
100 (metanol) |
5 |
|
Thuốc nhuộm azo (ví dụ Red disperse 1) |
|
|
|
|
|
50/50 (nước/CH3CN) |
0 |
5 |
100 (CH3CN) |
5 |
|
Tris(2.3- dibromopropyl) phosphat |
|
|
|
|
|
50/50 (nước/metanol) |
0 |
10 |
100 (metanol) |
5 |
|
Hợp chất clorin phenoxyaxit |
|
|
|
|
|
75/25 (A/metanol) |
2 |
15 |
40/60 (A/metanol) |
5 |
|
40/60 (A/metanol) |
3 |
5 |
75/25 (A/metanol) |
10 |
Trong đó A = 0,1 M amonium axetat (1 % axit axetic)
Cacbamat
Lụa chọn A
|
Thời gian (min) |
Pha động A (phần trăm) |
Pha động B (phần trăm) |
|
0 |
95 |
5 |
|
30 |
20 |
80 |
|
35 |
0 |
100 |
|
40 |
95 |
5 |
|
45 |
95 |
5 |
|
Trong đó A = 5 mM amonium axetat với 0,1 M axit axetic, và B = metanol Có cột tuỳ chọn thêm 0,5 M amonium axetat |
||
Lựa chọn B:
|
Thời gian (min) |
Pha động A (phần trăm) |
Pha động B (phần trăm) |
|
0 |
95 |
5 |
|
30 |
0 |
100 |
|
35 |
0 |
100 |
|
40 |
95 |
5 |
|
45 |
95 |
5 |
|
Trong đó A = nước có 0,1 M amonium axetat với 1 % axit axetic.
Có cột tuỳ chọn thêm 0,1 M amonium axetat. |
||
Bảng 2 – Các hợp chất thay đổi phổ khối lượng nhiệt phun
|
Thuốc nhuộm Disperse Azo Alkaloit Thuốc nhuộm Metyl Ure thơm Aromatic ureas Thuốc nhuộm Arylmetan Amid Thuốc nhuộm Coumarin Amin Thuốc nhuộm Anthraquinon Amino axit Thuốc nhuộm Xanten Hợp chất phospho hữu cơ Flame retardants Hợp chất axit phenoxy clo hoá Carbamat |
Bảng 3 – Giới hạn phát hiện và độ nhạy phương pháp của Red disperse 1 và caffein
|
Hợp chất |
Chế độ |
LOD pg |
EQL (7s) pg |
EQL (10s) pg |
|
Red disperse 1 |
SRM |
180 |
420 |
600 |
|
Quad đơn |
600 |
1400 |
2000 |
|
|
CAD |
2000 |
4700 |
6700 |
|
|
Caffein |
SRM |
45 |
115 |
150 |
|
Quad đơn |
84 |
200 |
280 |
|
|
CAD |
240 |
560 |
800 |
EQL: giới hạn định lượng ước lượng.
Bảng 4 – So sánh độ chuẩn và độ chụm của MS và MS/MS với HPLC/UV trong nước thử không có chất hữu cơ với Red disperse 1
|
Mẫu |
Phần trăm thu hồi |
|||
|
HLP/UV |
MS |
CAD |
SRM |
|
|
Thêm chuẩn 1 |
82,2 ± 0,2 |
92,5 ± 3,7 |
87,6 ± 4,6 |
95,5 ± 17,1 |
|
Thêm chuẩn 2 |
87,4 ± 0,6 |
90,2 ± 4,7 |
90.4 ± 9,9 |
90,0 ± 5.9 |
|
RPD |
6,1 % |
2,5 % |
3,2 % |
5,9 % |
Dữ liệu trong tài liệu tham khảo 16.
Bảng 5 – So sánh độ chuẩn và độ chụm của MS và MS/MS với HPLC/UV trong nước thải đô thị với Red disperse 1
|
Mẫu |
Phần trăm thu hồi |
||
|
HLP/UV |
MS |
CAD |
|
|
Thêm chuẩn 1 |
93,4 ± 0,3 |
102,0 ± 31 |
82,7 ± 13 |
|
Thêm chuẩn 2 |
96,2 ± 0,1 |
79,7 ± 5 |
83,7 ± 5,2 |
|
RPD |
3,0 % |
25% |
1,2% |
Số liệu trong tài liệu tham khảo 16.
Bảng 6 – Kết quả phân tích bùn hoạt hoá trong xử lý nước thải
|
Thu hồi Red disperse 1 (mg/L) |
|||
|
Mẫu |
HLP/UV |
MS |
CAD |
|
5 mg/L thêm chuẩn Nồng độ |
|||
|
1 |
0,721 ± 0,003 |
0,664 ± 0,030 |
0,796 ± 0,008 |
|
1-D |
0,731 ± 0,021 |
0,600 ± 0,068 |
0,768 ± 0,093 |
|
2 |
0,279 ± 0,000 |
0,253 ± 0,052 |
0,301 ± 0,042 |
|
3 |
0,462 ± 0,001 |
0,449 ± 0,016 |
0,510 ± 0.091 |
|
RPD |
1,3% |
10,1% |
3,6 % |
|
0 mg/L thêm chuẩn Nồng độ |
|||
|
1 |
0,000 |
0,005 ± 0,0007 |
0,001 |
|
1 – D |
0,000 |
0,006 ± 0,001 |
0,001 |
|
2 |
0,000 |
0,002 ± 0,0003 |
0,001 |
|
3 |
0,000 |
0,003 ± 0,0004 |
0,001 |
|
RPD |
– |
18,2 % |
– |
Số liệu trong tài liệu tham khảo 16
Bảng 7 – Khối lượng hiệu chuẩn và % mật độ tương đối của PEG 400
|
Khối lượng |
% mật độ tương đốia |
|
18,0 |
32,3 |
|
35,06 |
13,5 |
|
36,04 |
40,5 |
|
50,06 |
94,6 |
|
77,04 |
27,0 |
|
168,12 |
5,4 |
|
212,14 |
10,3 |
|
256,17 |
17,6 |
|
300,20 |
27,0 |
|
344,22 |
45,9 |
|
388,25 |
64,9 |
|
432,28 |
100 |
|
476,30 |
94,6 |
|
520,33 |
81,1 |
|
564,35 |
67,6 |
|
608,38 |
32,4 |
|
652,41 |
16,2 |
|
653,41 |
4,1 |
|
696,43 |
8,1 |
|
697,44 |
2,7 |
|
a cường độ được chuẩn về khối lượng 432. |
|
Bảng 8 – Khối lượng hiệu chuẩn và % mật độ tương đối của PEG 600
|
Sự phân bố cực đại |
% tương đối |
|
18,0 |
4,7 |
|
36,04 |
11,4 |
|
50,06 |
64,9 |
|
77,04 |
17,5 |
|
168,12 |
9,3 |
|
212,14 |
43,9 |
|
256,17 |
56,1 |
|
300,20 |
22,8 |
|
344,22 |
28,1 |
|
388,25 |
38,6 |
|
432,28 |
54,4 |
|
476,30 |
64,9 |
|
520,33 |
86,0 |
|
564,35 |
100 |
|
608,38 |
63,2 |
|
652,41 |
17,5 |
|
653,41 |
5,6 |
|
696,43 |
1,8 |
|
a cường độ được chuẩn về khối lượng 564 |
|
Bảng 9 – Thời gian duy trì và khối phổ nhiệt phun của các hợp chất phospho hữu cơ
|
Hợp chất |
Thời gian lưu (min) |
Khối phổ (% Sự phân bố tương đối)a |
|
Monocrotophos |
1:09 |
241 (100), 224 (14) |
|
Trichlorfon |
1:22 |
274(100), 257(19), 238(19) |
|
Dimethoate |
1:28 |
230 (100), 247 (20) |
|
Dichlorvos |
4:40 |
238 (100), 221 (40) |
|
Naled |
9:16 |
398 (100), 381 (23), 238 (5), 221 (2) |
|
Fensulfothion |
9:52 |
326(10), 309(100) |
|
Methyl parathion |
10:52 |
281 (100), 264 (8), 251 (21), 234 (48) |
|
Phorate |
13:30 |
278(4), 261 (100) |
|
Disulfoton |
13:55 |
292(10), 275(100) |
|
Merphos |
18:51 |
315(100), 299(15) |
|
a Với các phân tử chứa Cl, Br và S chỉ thống kê các pic cơ bản của nhôm đồng vị. |
||
Số liệu trong tài liệu tham khảo 17.
Bảng 10 – Độ chuẩn và giới hạn phát hiện các hợp chất phosphor hữu cơ chuẩn
|
Hợp chất |
lon |
Nồng độ chất lượng chuẩn (mg/L) |
% RSD |
MDL (ng) |
|
Dichlorvos |
238 |
2,0 |
16,0 |
4 |
|
12,5 |
13,0 |
|||
|
25,0 |
5,7 |
|||
|
50,0 |
4,2 |
|||
|
Dimethoat |
230 |
2,0 |
2,2 |
2 |
|
12,5 |
4,2 |
|||
|
25,0 |
13,0 |
|||
|
50,0 |
7,3 |
|||
|
Phorat |
261 |
2,0 |
0,84 |
2 |
|
12,5 |
14,0 |
|||
|
25,0 |
7,1 |
|||
|
50,0 |
4,0 |
|||
|
Disulfoton |
275 |
2,0 |
2,2 |
1 |
|
12,5 |
14,0 |
|||
|
25,0 |
6,7 |
|||
|
50,0 |
3,0 |
|||
|
Fensufothion |
309 |
2,0 |
4,1 |
0,4 |
|
12,5 |
9,2 |
|||
|
25,0 |
9,8 |
|||
|
50,0 |
2,5 |
|||
|
Naled |
398 |
2 |
9,5 |
0,2 |
|
12,5 |
9,6 |
|||
|
25,0 |
5,2 |
|||
|
50,0 |
6,3 |
|||
|
Merphos |
299 |
2 |
5,5 |
1 |
|
12,5 |
17 |
|||
|
25 |
3,9 |
|||
|
50 |
5,3 |
|||
|
Methyl Parathion |
281 |
2 |
– |
30 |
|
12,5 |
7,1 |
|||
|
25 |
4,8 |
|||
|
50 |
1,5 |
Số liệu trong tài liệu tham khảo 17.
Bảng 11 – Độ chính xác và độ chuẩn của nước uống nồng độ thấp (A), đất nồng độ thấp (B), nước uống nồng độ trung bình (C), trầm tích nồng độ trung bình (C)
|
Hợp chất |
Độ thu hồi trung bình (%) |
Độ lệch chuẩn |
Khối lượng |
Khoảng thu hồi (%) |
Số mẫu phân tích |
|
A |
|
|
mg/I |
|
|
|
Dimethoat |
70 |
7,7 |
5 |
85-54 |
15 |
|
Dichlorvos |
40 |
12 |
5 |
64-14 |
15 |
|
Naled |
0,5 |
1,0 |
5 |
2-0 |
15 |
|
Fensulfothion |
112 |
3,3 |
5 |
119-106 |
15 |
|
Methyl parathion |
50 |
28 |
10 |
105-0 |
15 |
|
Phorat |
16 |
35 |
5 |
86-0 |
15 |
|
Disulfoton |
3,5 |
8 |
5 |
19-0 |
15 |
|
Merphos |
237 |
25 |
5 |
287-187 |
15 |
|
B |
|
|
mg/g |
|
|
|
Dimethoat |
16 |
4 |
50 |
24-7 |
15 |
|
Dichforvos |
Không phát hiện được |
|
50 |
|
15 |
|
Naled |
Không phát hiện được |
|
50 |
|
15 |
|
Fensulfothion |
45 |
5 |
50 |
56-34 |
15 |
|
Methyl parathion |
Không phát hiện được |
|
100 |
|
15 |
|
Phorat |
78 |
15 |
50 |
109-48 |
15 |
|
Disulfoton |
36 |
7 |
50 |
49-22 |
15 |
|
Merphos |
118 |
19 |
50 |
155-81 |
15 |
|
C |
|
|
mg/l |
|
|
|
Dimethoat |
52 |
4 |
50 |
61-43 |
12 |
|
Dichlorvos |
146 |
29 |
50 |
204-89 |
12 |
|
Naled |
4 |
3 |
50 |
9-0 |
12 |
|
Fensulfothion |
65 |
7 |
50 |
79-51 |
12 |
|
Methyl parathion |
85 |
24 |
100 |
133-37 |
12 |
|
Phorat |
10 |
15 |
50 |
41-0 |
12 |
|
Disulfoton |
2 |
1 |
50 |
4-0 |
12 |
|
Merphos |
101 |
13 |
50 |
126-75 |
12 |
|
D |
|
|
mg/kg |
|
|
|
Dimethoat |
74 |
8,5 |
2 |
91-57 |
15 |
|
Dichlorvos |
166 |
25 |
2 |
216-115 |
15 |
|
Naled |
Không phát hiện được |
|
2 |
|
15 |
|
Fensulfothion |
72 |
8,6 |
2 |
90-55 |
15 |
|
Methyl parathion |
84 |
9 |
3 |
102-66 |
15 |
|
Phorat |
58 |
6 |
2 |
70-46 |
15 |
|
Disulfoton |
56 |
5 |
2 |
66-47 |
15 |
|
Merphos |
78 |
4 |
2 |
86-70 |
12 |
Số liệu trong tài liệu tham khảo 17
Bảng 12 – Độ chính xác và độ chuẩn của nước thải đô thị (A), nước uống (B), bùn thải hoá chất (C)
|
Hợp chất |
Độ thu hồi trung bình (%) |
Độ lệch chuẩn |
Lượng thêm chuẩn (ng/mL) |
Khoảng % thu hồi |
Số mẫu phân tích |
|
Tris-BP (A) |
25 |
8,0 |
2 |
41-9,0 |
15 |
|
(B) |
40 |
5,0 |
2 |
50-30 |
12 |
|
(C) |
63 |
11 |
100 |
84-42 |
8 |
Số liệu trong tài liệu tham khảo 18.
Bảng 13 – Vận hành đơn EQL cho TRIS-BP
|
Nồng độ (ng/mL) |
Diện tích trung bình |
Độ lệch chuẩn |
3* độ lệch chuẩn |
7* độ lệch chuẩn |
10* độ lệch chuẩn |
|
50 |
2675 |
782 |
2347 |
5476 |
7823 |
|
100 |
5091 |
558 |
|
|
|
|
150 |
7674 |
2090 |
|
|
|
|
200 |
8379 |
2030 |
|
|
|
|
LOD (ng/mL) |
EQL dưới (ng/mL) |
EQL trên (ng/mL) |
|
33 |
113 |
172 |
EQL: Giới hạn định lượng ước lượng
Số liệu trong tài liệu tham khảo 18.
Bảng 14 – Giới hạn phát hiện trong chế độ ion âm và ion dương đối với thuốc trừ cỏ clorin phenoxyaxit và bốn este
|
|
Chế độ ion dương Định lượng LOD |
Chế độ ion âm Định lượng LOD |
||
|
Hợp chất |
Ion |
(ng) |
Ion |
(ng) |
|
Dalapon |
Không phát hiện được |
|
141 (M–H)– |
11 |
|
Dicamba |
238 (M+NH4)+ |
13 |
184 (M–HCl)– |
3,0 |
|
2,4-D |
238 (M+NH4)+ |
2,9 |
184 (M–HCl)– |
50 |
|
MCPA |
218 (M+NH4)+ |
120 |
199 (M–1)– |
28 |
|
Diclorprop |
252 (M+NH4)+ |
2,7 |
235 (M–1)– |
25 |
|
MCPP |
232 (M+NH4)+ |
5,0 |
213 (M–1)– |
12 |
|
2,4,5-T |
272 (M+NH4)+ |
170 |
218 (M–HCl)– |
6,5 |
|
2,4,5-TP (Silvex) |
286 (M+NH4)+ |
160 |
269 (M–1)– |
43 |
|
Dinoseb |
228 (M+NH4–NO)+ |
24 |
240 (M)– |
19 |
|
2,4-DB |
266 (M+NH4)+ |
3,4 |
247 (M–1)– |
110 |
|
2,4-D.Butoxy etanol este |
321 (M+H)+ |
1,4 |
185 (M–C6H13O1)– |
|
|
2,4,5-T.Butoxy etanol este |
372 (M+NH4)+ |
0,6 |
195 (M–C6H15O3)– |
|
|
2,4,5-T.Butyl este |
328 (M+NH4)+ |
8,6 |
195 (M–C6H11O2)– |
|
|
2,4-D. etyl- hexyl este |
350 (M+NH4)+ |
1,2 |
161 ((M–C10H19O3)– |
|
Số liệu trong Tài liệu tham khảo 19.
Bảng 15 – Độ chính xác và độ chuẩn của phép thử đơn phòng thí nghiệm của các thuốc trừ cỏ axit phenolxy clo hoá
|
Hợp chất |
Độ thu hồi trung bìnha (%) |
Độ lệch chuẩn |
Lượng thêm chuẩn |
Khoảng thu hồi (%) |
Số mẫu phân tích |
|
|
|
Nước uống mức thấp |
mg/L |
|
|
||
|
Dicamba |
63 |
22 |
5 |
86-33 |
9 |
|
|
2,4-D |
26 |
13 |
5 |
37-0 |
9 |
|
|
MCPA |
60 |
23 |
5 |
92-37 |
9 |
|
|
MCPP |
78 |
21 |
5 |
116-54 |
9 |
|
|
Dichlorprop |
43 |
18 |
5 |
61-0 |
9 |
|
|
2,4,5-T |
72 |
31 |
5 |
138-43 |
9 |
|
|
Silvex |
62 |
14 |
5 |
88-46 |
9 |
|
|
2,4-DB |
29 |
24 |
5 |
62-0 |
9 |
|
|
Dinoseb |
73 |
11 |
5 |
85-49 |
9 |
|
|
Dalapon |
ND |
ND |
5 |
ND |
9 |
|
|
2,4-D, ester |
73 |
17 |
5 |
104-48 |
9 |
|
|
|
Nước uống mức cao |
mg/L |
|
I |
||
|
Dicamba |
54 |
30 |
50 |
103-26 |
9 |
|
|
2,4-D |
60 |
35 |
50 |
119-35 |
9 |
|
|
MCPA |
67 |
41 |
50 |
128-32 |
9 |
|
|
MCPP |
66 |
33 |
50 |
122-35 |
9 |
|
|
Dichlorprop |
66 |
33 |
50 |
116-27 |
9 |
|
|
2,4,5 – T |
61 |
23 |
50 |
99-44 |
9 |
|
|
Silvex |
74 |
35 |
50 |
132-45 |
9 |
|
|
2,4-DB |
83 |
25 |
50 |
120-52 |
9 |
|
|
Dinoseb |
91 |
10 |
50 |
102-76 |
9 |
|
|
Dalapon |
43 |
9,6 |
50 |
56-31 |
9 |
|
|
2,4-D. ester |
97 |
19 |
50 |
130-76 |
9 |
|
|
Cát mức thấp |
mg/g |
|
||||
|
Dicamba |
117 |
26 |
0,1 |
147-82 |
10 |
|
|
2,4-D |
147 |
23 |
0,1 |
180-118 |
10 |
|
|
MCPA |
167 |
79 |
0,1 |
280-78 |
10 |
|
|
MCPP |
142 |
39 |
0,1 |
192-81 |
10 |
|
|
Dichlorprop |
ND |
ND |
0,1 |
ND |
10 |
|
|
2,4,5-T |
134 |
27 |
0,1 |
171-99 |
10 |
|
|
Silvex |
121 |
23 |
0,1 |
154-85 |
10 |
|
|
2,4-DB |
199 |
86 |
0,1 |
245-0 |
10 |
|
|
Dinoseb |
76 |
74 |
0,1 |
210-6 |
10 |
|
|
Dalapon |
ND |
ND |
0,1 |
ND |
10 |
|
|
2,4-D.ester |
180 |
58 |
0,1 |
239-59 |
7 |
|
|
|
Cát mức cao |
mg/L |
|
|
||
|
Dicamba |
153 |
33 |
1 |
209-119 |
9 |
|
|
2,4-D |
218 |
27 |
1 |
276-187 |
9 |
|
|
MCPA |
143 |
30 |
1 |
205-111 |
9 |
|
|
MCPP |
158 |
34 |
1 |
226-115 |
9 |
|
|
Dichlorprop |
92 |
37 |
1 |
161-51 |
9 |
|
|
2,4,5-T |
160 |
29 |
1 |
204-131 |
9 |
|
|
Silvex |
176 |
34 |
1 |
225-141 |
9 |
|
|
2,4-DB |
145 |
22 |
1 |
192-110 |
9 |
|
|
Dinoseb |
114 |
28 |
1 |
140-65 |
9 |
|
|
Dalapon |
287 |
86 |
1 |
418-166 |
9 |
|
|
2,4-D. ester |
20 |
3,6 |
1 |
25-17 |
7 |
|
|
|
Tro của đô thị mức thấp |
mg/g |
|
|
||
|
Dicamba |
83 |
22 |
0,1 |
104-48 |
9 |
|
|
2,4-D |
ND |
ND |
0,1 |
ND |
9 |
|
|
MCPA |
ND |
ND |
0,1 |
ND |
9 |
|
|
MCPP |
ND |
ND |
0,1 |
ND |
9 |
|
|
Dichlorprop |
ND |
ND |
0,1 |
ND |
9 |
|
|
2,4,5-T |
27 |
25 |
0,1 |
60-0 |
9 |
|
|
Silvex |
68 |
38 |
0,1 |
128-22 |
9 |
|
|
2,4-DB |
ND |
ND |
0,1 |
ND |
9 |
|
|
Dinoseb |
44 |
13 |
0,1 |
65-26 |
9 |
|
|
Dalapon |
ND |
ND |
0,1 |
ND |
9 |
|
|
2,4-D. ester |
29 |
23 |
0,1 |
53-0 |
6 |
|
|
|
Tro của đô thị mức cao |
mg/g |
|
|
||
|
Dicamba |
66 |
21 |
1 |
96-41 |
9 |
|
|
2,4-D |
8,7 |
4,8 |
1 |
21-5 |
9 |
|
|
MCPA |
3,2 |
4,8 |
1 |
10-0 |
9 |
|
|
MCPP |
10 |
4,3 |
1 |
16-4,7 |
9 |
|
|
Dichlorprop |
ND |
ND |
1 |
ND |
9 |
|
|
2,4,5-T |
2,9 |
1,2 |
1 |
3,6-0 |
9 |
|
|
Silvex |
6,0 |
3,1 |
1 |
12-2,8 |
9 |
|
|
2,4-DB |
ND |
ND |
1 |
ND |
9 |
|
|
Dinoseb |
16 |
6,8 |
1 |
23-0 |
9 |
|
|
Dalapon |
ND |
ND |
1 |
ND |
9 |
|
|
2,4-D. ester |
1,9 |
1,7 |
1 |
6,7-0 |
6 |
|
|
a Độ thu hồi tổng số là của chế độ ion âm, trừ trường hợp ete 2,4-D ND: Không phát hiện được |
||||||
Bảng 16 – Dữ liệu độ chính xác và độ chuẩn của phép thử liên phòng thí nghiệm đối với thuốc trừ cỏ axit phenoxy clo hoá
|
Hợp chất |
Nồng độ thêm chuẩn |
Giá trị trung bình (% Độ thu hồi)a |
% độ lệch chuẩn tương đối b |
|
|
|
|
500 mg/L |
|
|
|
2,4,5-T |
|
90 |
23 |
|
|
2,4,5-T, butoxy |
|
90 |
29 |
|
|
2,4-D |
|
86 |
17 |
|
|
2,4-DB |
|
95 |
22 |
|
|
Dalapon |
|
83 |
13 |
|
|
Dicamba |
|
77 |
25 |
|
|
Dichlorprop |
|
84 |
20 |
|
|
Dinoseb |
|
78 |
15 |
|
|
MCPA |
|
89 |
11 |
|
|
MCPP |
|
86 |
12 |
|
|
Silvex |
|
96 |
27 |
|
|
|
|
50 mg/L |
|
|
|
2,4,5-T |
|
62 |
68 |
|
|
2,4,5-T, butoxy |
|
85 |
9 |
|
|
2,4-D |
|
64 |
80 |
|
|
2,4-DB |
|
104 |
28 |
|
|
Dalapon |
|
121 |
99 |
|
|
Dicamba |
|
90 |
23 |
|
|
Dichlorprop |
|
96 |
15 |
|
|
Dinoseb |
|
86 |
57 |
|
|
MCPA |
|
96 |
20 |
|
|
MCPP |
|
76 |
74 |
|
|
Silvex |
|
65 |
71 |
|
|
|
|
5 mg/L |
|
|
|
2,4,5-T |
|
90 |
28 |
|
|
2,4,5-T, butoxy |
|
99 |
17 |
|
|
2,4-D |
|
103 |
31 |
|
|
2,4-DB |
|
96 |
21 |
|
|
Dalapon |
|
150 |
4 |
|
|
Dicamba |
|
105 |
12 |
|
|
Dichlorprop |
|
102 |
22 |
|
|
Dinoseb |
|
108 |
30 |
|
|
MCPA |
|
94 |
18 |
|
|
MCPP |
|
98 |
15 |
|
|
Silvex |
|
87 |
15 |
|
|
a Giá trị trung bình kết quả thí nghiệm phân tích hai lần của ba phòng thí nghiệm. b % RSD kết quả thí nghiệm phân tích hai lần của ba phòng thí nghiệm. |
||||
Số liệu trong tài liệu tham khảo 20.
Bảng 17 – So sánh LOD: Phương pháp 8151 với Phương pháp 8321
|
Hợp chất ion hoá |
Phương pháp 8151 Mẫu nước GC/ECD EDL (mg/L)a |
Phương pháp 8321 Mẫu nước HPLC/MS/TS LOD (mg/L) |
Chế độ |
|
Dalapon |
1,3 |
1,1 |
(-) |
|
Dicamba |
0,081 |
0,3 |
(-) |
|
2,4-D |
0,2 |
0,29 |
(+) |
|
MCPA |
0,056b |
2,8 |
(-) |
|
Dichlorprop |
0,26 |
0,27 |
(+) |
|
MCPP |
0,09 |
0,50 |
(+) |
|
2,4,5-T |
0,08 |
0,65 |
(-) |
|
2,4,5-TP (Silvex) |
0,075 |
4,3 |
(–) |
|
2,4-DB |
0,8 |
0,34 |
(+) |
|
Dinoseb |
0,19 |
1,9 |
(-) |
|
a EDL Giới hạn phát hiện ước tính, được quy định như MDL hoặc nồng độ chất phân tích trong mẫu thu được một pic trong dịch chiết cuối cùng có tỉ số giữa độ nhiễu và tín hiệu xấp xỉ 5. b 40 CFR Phần 136, Phụ lục B (49 FR 43234). Sắc ký dùng cột mao quản lõi dây. |
|||
Bảng 18 – Xác định giới hạn phát hiện phương pháp đơn phòng thí nghiệm và kết quả độ chụm – nướcc
|
Chất phân tích |
Độ thu hồi trung bình % |
Độ lệch chuẩn |
% RSD |
MDLb mg/L |
|
Aldicarb sulfoxida |
7,5 |
0,27 |
72,4 |
0,8 |
|
Aldicarb sulfon |
88,4 |
0,44 |
50,3 |
1,3 |
|
Oxamyl |
60,7 |
0,10 |
16,6 |
0,3 |
|
Methomyl |
117 |
0,49 |
41,5 |
1,5 |
|
3-Hydroxycarbofurana |
37,4 |
0,25 |
65,4 |
0,8 |
|
Fenuron |
104 |
0,20 |
19,3 |
0,6 |
|
Benomyl/Carbendazim |
67,3 |
0,13 |
19,7 |
0,4 |
|
Aldicarb |
93,7 |
0,46 |
49,6 |
1,4 |
|
Aminocarb |
117 |
0,53 |
44,9 |
1,6 |
|
Carbofuran |
94,2 |
0,17 |
17,7 |
0,5 |
|
Propoxur |
106 |
0,32 |
30,4 |
1,0 |
|
Monuron |
95,6 |
0,24 |
25,6 |
0,7 |
|
Bromacil |
86,4 |
0,12 |
14,1 |
0,4 |
|
Tebuthiuron |
106 |
0,17 |
16,1 |
0,5 |
|
Carbaryl |
85,1 |
0,29 |
34,1 |
0,9 |
|
Fluometuron |
89,1 |
0,19 |
21,7 |
0,6 |
|
Propham |
84,2 |
0,15 |
17,3 |
0,4 |
|
Propachlor |
98,5 |
0,16 |
16,0 |
0,5 |
|
Diuron |
95,6 |
0,14 |
14,7 |
0,4 |
|
Siduron |
105 |
0,27 |
25,9 |
0,8 |
|
Methiocarb |
92,4 |
0,16 |
17,5 |
0,5 |
|
Barban |
90,5 |
0,79 |
17,4 |
2,4 |
|
Linuron |
97,7 |
0,19 |
19,5 |
0,6 |
|
Chloropropham |
89,1 |
0,68 |
15,2 |
2,0 |
|
Mexacarbat |
80,0 |
1,41 |
35,1 |
4,2 |
|
Chloroxuron |
109 |
0,32 |
29,2 |
1,0 |
|
Neburon |
92,5 |
0,14 |
14,9 |
0,4 |
|
a Giá trị thu được từ tính toán hệ số tương ứng nội bộ. b Việc xác định giới hạn phát hiện của phương pháp được dựa trên 20 dịch chiết nước. Mức chuẩn thêm Aldicarb sulfoxit, barban, Cloropropham và mexacacba là 5 mg/L. Tất cả các chất phân tích khác được thêm chuẩn ở mức 1 mg/L. Giới hạn phát hiện của phương pháp được xác định khi nhân độ lệch chuẩn với 3. Việc định lượng được tiến hành sử dụng giá trị hồi qui tuyến tính trung bình, trừ khi có các chỉ định khác. |
||||
c Số liệu từ Tài liệu tham khảo 22.
Bảng 19 – Xác định giới hạn phát hiện phương pháp đơn phòng thí nghiệm và kết quả độ chụm – đấtb
|
Chất phân tích |
Độ thu hồi trung bình % |
Độ lệch chuẩn |
% RSD |
MDLa mg/g |
|
Aldicarb sulfoxid |
66,9 |
0,0492 |
58,9 |
0,15 |
|
Aldicarb sulfon |
118 |
0,0076 |
25,7 |
0,023 |
|
Oxamyl |
89,6 |
0,0049 |
21,9 |
0,015 |
|
Methomyl |
86,8 |
0,0051 |
23,6 |
0,015 |
|
3-Hydroxycarbofuran |
103 |
0,0116 |
45,0 |
0,035 |
|
Fenuron |
91,2 |
0,0049 |
21,6 |
0,015 |
|
Benomyl/Carbendazim |
68,0 |
0,0082 |
47,0 |
0,025 |
|
Aldicarb |
72,0 |
0,0056 |
30,1 |
0,017 |
|
Aminocarb |
84,4 |
0,0082 |
38,7 |
0,025 |
|
Carbofuran |
102 |
0,0083 |
32,7 |
0,025 |
|
Propoxur |
95,2 |
0,0091 |
38,2 |
0,027 |
|
Monuron |
107 |
0,0077 |
28,8 |
0,023 |
|
Bromacil |
99,6 |
0,0069 |
27,5 |
0,021 |
|
Tebuthiuron |
96,8 |
0,0071 |
29,5 |
0,021 |
|
Carbaryl |
99,6 |
0,0054 |
21,7 |
0,016 |
|
Fluometuron |
92,8 |
0,0035 |
15,1 |
0,011 |
|
Propham |
100 |
0,0039 |
15,7 |
0,012 |
|
Propachlor |
114 |
0,0037 |
13,0 |
0,011 |
|
Diuron |
101 |
0,0060 |
23,8 |
0,018 |
|
Siduron |
107 |
0,0063 |
23,7 |
0,019 |
|
Methiocarb |
124 |
0,0054 |
17,5 |
0,016 |
|
Barban |
108 |
0,0333 |
24,8 |
0,10 |
|
Linuron |
113 |
0,0037 |
13,0 |
0,011 |
|
Chloropropham |
104 |
0,0217 |
16,6 |
0,065 |
|
Mexacarbat |
62,2 |
0,0119 |
15,3 |
0,036 |
|
Chloroxuron |
97,6 |
0,0031 |
12,6 |
0,009 |
|
Neburon |
110 |
0,0044 |
16,0 |
0,011 |
|
a Việc xác định giới hạn phát hiện của phương pháp được dựa trên 20 dịch chiết đất. Mức chuẩn thêm Aldicarb sulfoxit, barban, Cloropropham và mexacacba là 0,125 mg/g. Tất cả các chất phân tích khác được thêm chuẩn ở mức 0,025 mg/g. Giới hạn phát hiện của phương pháp được xác định khi nhân độ lệch chuẩn với 3. Việc định lượng được tiến hành sử dụng giá trị hồi qui tuyến tính trung bình, trừ khi có các chỉ định khác. |
||||
b Số liệu từ Tài liệu tham khảo 22.
Bảng 20 – Đánh giá đơn phòng thí nghiệm về độ thu hồi trung bình và độ chụm – nướcc
|
Chất phân tích |
Độ thu hồi trung bìnhb % |
Độ lệch chuẩn |
% RSD |
|
Aldicarb sulfoxid |
7,6 |
2,8 |
37,0 |
|
Aldicarb sulfon |
56,0 |
27,1 |
48,5 |
|
Oxamyla |
38,9 |
17,9 |
45,9 |
|
Methomyl |
52,0 |
19,6 |
37,7 |
|
3-Hydroxycarbofuran |
22,2 |
9,3 |
41,7 |
|
Fenuron |
72,5 |
22,0 |
30,3 |
|
Benomyl/Carbendazim |
47,3 |
14,7 |
31,0 |
|
Aldicarb |
81,0 |
13,7 |
16,9 |
|
Aminocarb |
109 |
38,3 |
35,1 |
|
Carbofuran |
85,5 |
10,0 |
11,7 |
|
Propoxur |
79,1 |
13,7 |
17,3 |
|
Monuron |
91,8 |
11,3 |
12,3 |
|
Bromacil |
87,6 |
12,1 |
13,8 |
|
Tebuthiuron |
87,1 |
9,0 |
10,3 |
|
Carbaryl |
82,1 |
13,5 |
16,5 |
|
Fluometuron |
84,4 |
8,3 |
9,8 |
|
Propham |
80,7 |
13,8 |
17,1 |
|
Propachlor |
84,3 |
10,0 |
11,9 |
|
Diuron |
90,8 |
14,1 |
15,6 |
|
Siduron |
88,0 |
9,5 |
10,8 |
|
Methiocarb |
93,3 |
12,8 |
13,8 |
|
Barban |
88,1 |
11,2 |
12,7 |
|
Linuron |
87,1 |
16,8 |
19,3 |
|
Chloropropham |
94,9 |
15,3 |
16,1 |
|
Mexacarbat |
79,8 |
12,9 |
16,2 |
|
Chloroxuron |
106 |
24,9 |
23,5 |
|
Neburon |
85,3 |
12,6 |
14,8 |
|
a Giá trị thu được từ tính toán hệ số đáp trả nội bộ. b Tiến hành phân tích 9 mẫu đã thêm chuẩn với 3 mức nồng độ. Nồng độ của Aldicarb sulfoxit, barban, Cloropropham và mexacacba tương ứng là 25 mg/L, 50 mg/L và 100 mg/L. Tất cả các nồng độ phân tích khác ở mức 5 mg/L, 10 mg/L và 20 mg/L. Một mẫu vượt không coi như nằm ngoài. Tổng số mẫu đã phân tích là 26 mẫu. Việc định lượng được tiến hành sử dụng giá trị hồi qui tuyến tính trung bình, trừ khi có các chỉ định khác. |
|||
c Số liệu từ Tài liệu tham khảo 22.
Bảng 21 – Đánh giá đơn phòng thí nghiệm về độ thu hồi trung bình và độ chụm – Đấtb
|
Chất phân tích |
Độ thu hồi trung bìnhb % |
Độ lệch chuẩn |
% RSD |
|
Aldicarb sulfoxid |
66,9 |
31,3 |
46,7 |
|
Aldicarb sulfon |
162 |
51,4 |
31,7 |
|
Oxamyla |
78,9 |
46,1 |
58,5 |
|
Methomyl |
84,9 |
25,8 |
30,4 |
|
3-Hydroxycarbofuran |
105 |
36,3 |
34,5 |
|
Fenuron |
91,9 |
16,7 |
18,1 |
|
Benomyl/Carbendazim |
95,6 |
18,2 |
19,0 |
|
Aldicarb |
97,9 |
17,0 |
17,4 |
|
Aminocarb |
133 |
44,7 |
33,6 |
|
Carbofuran |
109 |
14,4 |
13,2 |
|
Propoxur |
104 |
16,5 |
15,9 |
|
Monuron |
101 |
12,4 |
12,3 |
|
Bromacil |
100 |
9,0 |
9,0 |
|
Tebuthiuron |
104 |
11,9 |
11,5 |
|
Carbaryl |
102 |
15,5 |
15,2 |
|
Fluometuron |
94,5 |
15,7 |
16,7 |
|
Propham |
92,8 |
12,0 |
12,9 |
|
Propachlor |
94,6 |
10,3 |
10,9 |
|
Diuron |
107 |
17,4 |
16,2 |
|
Siduron |
100 |
12,0 |
12,0 |
|
Methiocarb |
107 |
14,2 |
13,2 |
|
Barban |
92,3 |
15,6 |
16,9 |
|
Linuron |
104 |
13,6 |
13,1 |
|
Chloropropham |
105 |
9,3 |
8,9 |
|
Mexacarbat |
77,2 |
9,8 |
12,7 |
|
Chloroxuron |
121 |
27,3 |
22,5 |
|
Neburon |
92,1 |
16,5 |
17,9 |
|
a Tiến hành phân tích 9 mẫu đã thêm chuẩn với 3 mức nồng độ. Nồng độ của Aldicarb sulfoxit, barban, Cloropropham và mexacacba tương ứng là 0,625 mg/g; 1,250 mg/g và 2,5 mg/g. Tất cả các nồng độ phân tích khác ở mức 0,125 mg/g. 0,25 mg/g và 0,5 mg/g. Một mẫu vượt không coi nhu nằm ngoài. Tổng số mẫu đã phân tích là 26 mẫu. Việc định lượng được tiến hành sử dụng giá trị hồi qui tuyến tính trung bình, trừ khi có các chỉ định khác. |
|||
b Số liệu từ Tài liêu tham khảo 22
Bảng 22 – Đánh giá liên phòng thí nghiệm độ chính xác của phương pháp
(Sau khi loại bỏ phần tử ngoại biên)d
|
Chất phân tích |
Mẫu có nồng độ cao a |
Mẫu có nồng độ trung bìnhb |
Mẫu có nồng độ thấpc |
|
Aldicarb |
98,7 |
110 |
52,0 |
|
Bendiocarb |
81,4 |
95,0 |
52,0 |
|
Carbaryl |
92,0 |
108 |
62,0 |
|
Carbendazim |
125 |
138 |
128 |
|
Carbofuran |
87,8 |
92,3 |
72,0 |
|
Diuron |
79,9 |
98,8 |
66,0 |
|
Linuron |
84,8 |
93,0 |
82,0 |
|
Methomyl |
93,3 |
90,8 |
90,0 |
|
Oxamyl |
83,8 |
88,0 |
98,0 |
|
a Mỗi phòng thí nghiệm lặp lại 3 lần, tiến hành tại 8 – 9 phòng thí nghiệm (Bảng 26 của tài liệu tham khảo 23). Mỗi hợp chất có nồng độ chính xác là 90 mg/L, trừ trường hợp Carbendazim là 22,5 mg/L; b Mỗi phòng thí nghiệm lặp lại 2 lần, tiến hành tại 8 – 9 phòng thí nghiệm (Bảng 26 của tài liệu tham khảo 23). Mỗi hợp chất có nồng độ chính xác là 40 mg/L, trừ trường hợp Carbendazim là 10 mg/L; c Mỗi phòng thí nghiệm lặp lại 3 lần, tiến hành tại 8 – 9 phòng thí nghiệm (Bảng 26 của tài liệu tham khảo 23). Mỗi hợp chất có nồng độ chính xác là 5 mg/L, trừ trường hợp Carbendazim là 1,25 mg/L; |
|||
d Số liệu từ Tài liệu tham khảo 23.
Bảng 23 – Đánh giá liên phòng thí nghiệm về độ chính xác của phương pháp
(Sau khi loại bỏ phần tử ngoại biên)a
|
Chất phân tích |
Nồng độ cao |
Nồng độ trung bình |
Nồng độ thấp |
||||||||||||
|
Trung bình |
Sr |
SR |
% RSDR |
% RSDR |
Trung bình |
Sr |
SR |
% RSDr |
% RSDR |
Trung bình |
Sr |
SR |
% RSDr |
% RSDR |
|
|
Aldicarb |
88,8 |
11,4 |
34,4 |
12,9 |
38,8 |
44,1 |
7,7 |
17,0 |
17,5 |
38,5 |
2,6 |
0,9 |
2,6 |
33,1 |
98,2 |
|
Bendiocarb |
73,3 |
16,1 |
39,3 |
21,9 |
53,6 |
38,0 |
6,6 |
16,6 |
17,3 |
43,7 |
2,6 |
0,6 |
1,6 |
21,3 |
61,9 |
|
Carbaryl |
82,8 |
11,7 |
34,0 |
14,2 |
41,1 |
43,1 |
3,0 |
15,7 |
7,0 |
36,4 |
3,1 |
0,7 |
2,3 |
23,3 |
75,8 |
|
Carbendazim |
28,1 |
5,6 |
15,3 |
19,9 |
54,4 |
13,8 |
1,4 |
8,9 |
10,4 |
64,2 |
1,6 |
0,4 |
1,1 |
26,1 |
68,2 |
|
Carbofuran |
79,0 |
16,7 |
35,2 |
21,2 |
44,5 |
36,9 |
5,0 |
16,3 |
13,6 |
44,3 |
3,6 |
0,9 |
3,3 |
25,2 |
91,6 |
|
Diuron |
71,9 |
13,1 |
26,1 |
18,2 |
36,3 |
39,5 |
2,6 |
11,8 |
6,5 |
29,8 |
3,3 |
0,5 |
2,6 |
16,2 |
77,9 |
|
Linuron |
76,3 |
8,3 |
32,5 |
10,9 |
42,6 |
37,2 |
3,9 |
13,4 |
10,5 |
35,9 |
4,1 |
0,6 |
2,1 |
15,7 |
51,4 |
|
Methomyl |
84,0 |
10,8 |
29,4 |
12,9 |
35,0 |
36,3 |
2,8 |
15,0 |
7,8 |
41,2 |
4,5 |
0,7 |
4,1 |
15,3 |
92,9 |
|
Oxamyl |
75,5 |
12,4 |
37,0 |
16,4 |
49,1 |
35,2 |
3,7 |
20,8 |
10,4 |
59,1 |
4,9 |
0,5 |
4,6 |
9,7 |
93,6 |
|
Trung bình |
|
|
|
16,5 |
43,9 |
|
|
|
11,2 |
43,7 |
|
|
|
20,7 |
79,1 |
|
Độ lệch chuẩn |
|
|
|
4,0 |
7,1 |
|
|
|
4,1 |
11,2 |
|
|
|
7,1 |
16,3 |
Sr và SR là độ lệch chuẩn của độ lặp lại và độ tái lập, tương ứng. RSDr và RSDR là độ lệch chuẩn tương đối của độ lặp lại và độ tái lập, tương ứng. Đơn vị của giá trị trung bình sr và SR là mg/L.
a Số liệu từ Tài liệu tham khảo 23.
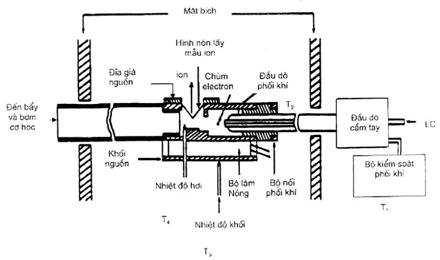
Hình 1 – Sơ đồ đầu dò nhiệt phun và nguồn ion
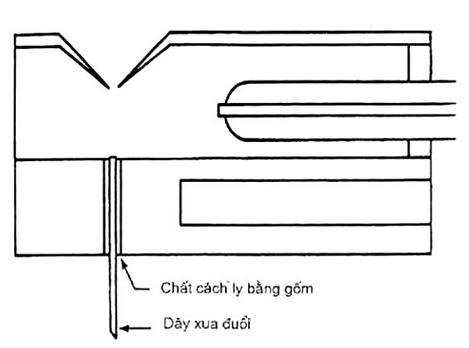
Hình 2 – Nguồn phun nhiệt có dây xua đuổi (cấu hình độ nhạy cao)
Hình 3 – Nguồn phun nhiệt có dây xua đuổi (cấu hình CAD)
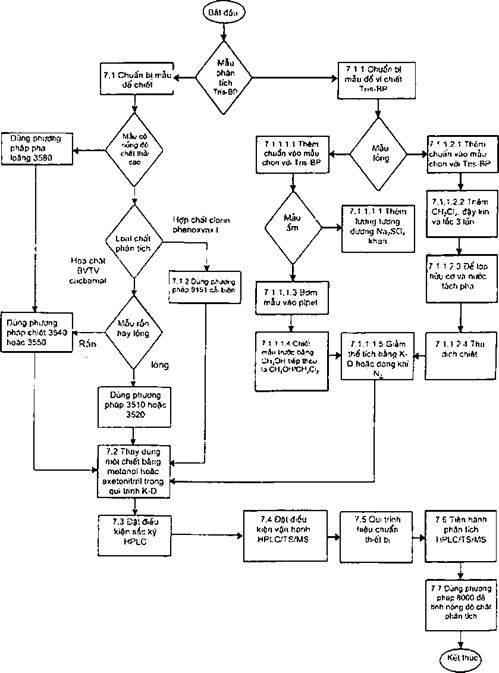
Hình 4 – Lưu đồ quy trình
THƯ MỤC TÀI LIỆU THAM KHẢO
[1] Voyksner. R.D., Haney. C.A., “Optimization and Application of Thermospray High-Performance Liquid Chromatography/Mass Spectrometry”. Anal. Chem., 1985, 57, 991-996
[2] Blakley, C.R., Vestal, M.L., “Thermospray Interpace for Liquid Chromatography/Mass Spectrometry” Anal Chem., 1983, 55, 750-754.
[3] Taylor, V., Hickey, D.M., Marsden, P.J “Single Laboratory Validation of EPA Method 8140“ EPA- 600/4-87/009, U.S. Environmental Protection Agency. Las Vegas, NV, 1987, 144 pp
[4] “Guidelines for Data Acquisition and Data quality Evaluation in Environmental Chemistry”, Anal, Chem., 1980, 52, 2242-2249.
[5] Betowski. L.D., Jones. T.L., ‘‘The Analysis of Organophosphorus Pesticide Samples by HPLC/MS and HPLC/MS/MS”, Environmental Science and Technology, 1988.
[8] U.S. EPA 2nd Annual Report on Carcinogens. NTP 81-43, Dec. 1981, pp. 236-237.
[9] Blum, A., Ames, B.N., Science 195, 1997, 17.
[10] Zweidinger, R.A., Cooper, S.D., Pellazari. E.D., Measurements of Organic Pollutants in Water and Wastewater, ASTM 686.
[11] Cremlyn. R., Pesticides: Preparation and mode of Action, John Wiley and Sons, Chichester, 1978, p. 142
[12] Cotterill, E.G., Byast, T.H., “HPLC of Pesticide Residues in Environmental Sample”, in Liquid Chromatography in Environmental Analysis, Laurence, J.F., Ed., Humana Press, Clifton. NJ, 1984.
[13] Voyksner, R.D., “Thermospray HPLC/MS for Monitoring the Environment” in Applications of New Mass Spectrometry Techniques in Pesticide Chemistry: Rosen, J.D., Ed., John Wiley and Sons. New York. 1987.
[14| Yinon, J., Jones, T.L., Betovvski, L.D., Rap. Comm. Mass Spectrom., 1989. 3. 38.
[15] Shore, F.L., Amick, E.N., Pan, S.T., Gurka, D.F., “Single Laboratory Validation of EPA Method 8150 for the Analysis of Chlorinated Herbicides in Hazadous Waste“, EPA/600/4-85/060. U.S. Environmental Protection Agency, Las Vegas, NV, 1985
[16] “Development and Evaluations of an LC/MS/MS Protocol”, EPA/600/X-86/328, Dec. 1986.
[17] “An LC/MS Performance Evaluation Study of Organophosphorus Pesticide” EPA/600/X-89/006, Jan.1989
[18] “A Performance Evaluation Study of a Liquid Chromatography/Mass Spectrometry Method for Tris- (2,3-Dibromopropyl) Phosphate”, EPA/600/X-89/135, June 1989.
[19] “Liquid Chromatography/Mass Spectrometry Performance Evaluation of Chlorinated Phenoxyacid Herbicide and Their Esters”, EPA/600/X-89/176, July 1989.
[20] “An Interlaboratory Comparison of an SW-846 Method for the Analysis of the Chlorinated Phenoxyacid Herbicides by LC/MS” EPA/600/X-90/133, June 1990.
[21] Somasundaram, L., and J.R. Coates, Ed., “Pesticide Transformation Products Fate and Significance in the Environment”, ACS Symposium Series 495, Ch.13. 1991.
[22] Single-Laboratory Evaluation of Carbamates, APPL, Inc., Fresno, CA.
[23] “Interlaboratory Calibration Study of a Thermospray-Liquid Chromatography/Mass Spectrometry (TS-LC/MS) Method for Selected Carbamate Pesticide”, EPA/600/X-92/102, August 1992.
