
Nội dung toàn văn Tiêu chuẩn quốc gia TCVN 6325:2013 (ASTM D 664-11a) về Sản phẩm dầu mỏ – Xác định trị số axit – Phương pháp chuẩn độ điện thế
TIÊU CHUẨN QUỐC GIA
TCVN 6325:2013
ASTM D 664-11a
SẢN PHẨM DẦU MỎ – XÁC ĐỊNH TRỊ SỐ AXIT – PHƯƠNG PHÁP CHUẨN ĐỘ ĐIỆN THẾ
Standard Test Method for Acid Number of Petroleum Products by Potentiometric Titration
Lời nói đầu
TCVN 6325:2013 thay thế TCVN 6325:2007.
TCVN 6325:2013 được xây dựng trên cơ sở hoàn toàn tương đương với ASTM D 664-11a Standard Test Method for Acid Number of Petroleum Products by Potentiometric Titration với sự cho phép của ASTM quốc tế, 100 Barr Harbor Drive, West Conshohocken, PA 19428, USA. Tiêu chuẩn ASTM D 664-11a thuộc bản quyền của ASTM quốc tế.
TCVN 6325:2013 do Tiểu ban kỹ thuật tiêu chuẩn quốc gia TCVN/TC28/SC2 Nhiên liệu lỏng – Phương pháp thử biên soạn, Tổng cục Tiêu chuẩn Đo lường Chất lượng đề nghị, Bộ Khoa học và Công nghệ công bố.
SẢN PHẨM DẦU MỎ – XÁC ĐỊNH TRỊ SỐ AXIT – PHƯƠNG PHÁP CHUẨN ĐỘ ĐIỆN THẾ
Standard test method for acid number of petroleum products by potentiometric titration
1. Phạm vi áp dụng
1.1. Tiêu chuẩn này quy định phương pháp xác định thành phần axit trong sản phẩm dầu mỏ, dầu bôi trơn, điêzen sinh học và hỗn hợp điêzen sinh học.
1.1.1. Phương pháp thử A: áp dụng đối với sản phẩm dầu mỏ và dầu bôi trơn có thể hòa tan hoặc gần hòa tan trong hỗn hợp của toluen và propan-2-ol. Phương pháp này có thể áp dụng để xác định các axit có hằng số phân ly trong nước lớn hơn 10–9; axit cực yếu có hằng số phân ly nhỏ hơn 10-9 không gây cản trở. Các muối sẽ phản ứng nếu hằng số thủy phân lớn hơn 10–9. Dải trị số axit bao gồm trong công bố về độ chụm là từ 0,1 mg/g KOH đến 150 mg/g KOH.
1.1.2. Phương pháp thử B: được xây dựng riêng cho điêzen sinh học và hỗn hợp điêzen sinh học có độ axit thấp và khó tan. Phương pháp thử này yêu cầu sử dụng chuẩn độ tự động có khả năng tự động tìm kiếm điểm cuối.
CHÚ THÍCH 1: Trong các dầu bôi trơn mới và dầu đã sử dụng, các thành phần có tính axit bao gồm các axit vô cơ và hữu cơ, các este, phenol, lacton, chất nhựa, muối của các kim loại nặng, muối amoni, và muối của bazơ yếu khác, các muối axit của các axit đa chức và các phụ gia như chất ức chế và chất tẩy rửa.
1.2. Phương pháp này cũng được áp dụng để phát hiện sự thay đổi tương đối của dầu nhờn trong quá trình sử dụng dưới điều kiện oxy hóa, không kể tới màu sắc và các tính chất khác của dầu đã bị oxy hóa. Mặc dù việc chuẩn độ được tiến hành dưới những điều kiện cân bằng xác định, phương pháp này không dự kiến để đo độ axit tuyệt đối dùng để dự đoán tính năng của dầu nhờn trong điều kiện sử dụng. Không có mối liên hệ chung nào giữa độ ăn mòn ổ đỡ với trị số axit.
CHÚ THÍCH 2: Trị số axit thu được trong tiêu chuẩn này có thể bằng hoặc không bằng như trị số axit thu được theo phương pháp thử trong TCVN 2695 (ASTM D 974) và ASTM D 3339. Hiện nay chưa có sự so sánh tương quan của phương pháp này với các phương pháp không chuẩn độ khác.
CHÚ THÍCH 3: Một số phòng thử nghiệm có kết quả khác nhau khi dùng phương pháp thử ASTM D 664 sử dụng chất đệm có nước và không có nước.
1.3. Các giá trị tính theo hệ SI là giá trị tiêu chuẩn.
1.4. Tiêu chuẩn này không đề cập đến tất cả các vấn đề liên quan đến an toàn khi sử dụng. Người sử dụng tiêu chuẩn này có trách nhiệm thiết lập các nguyên tắc về an toàn và bảo vệ sức khỏe cũng như khả năng áp dụng phù hợp với các giới hạn quy định trước khi đưa vào sử dụng.
2. Tài liệu viện dẫn
Các tài liệu viện dẫn sau là cần thiết khi áp dụng tiêu chuẩn. Đối với các tài liệu viện dẫn ghi năm công bố thì áp dụng bản được nêu. Đối với các tài liệu viện dẫn không ghi năm công bố thì áp dụng phiên bản mới nhất, bao gồm cả các bản sửa đổi (nếu có).
TCVN 2117 (ASTM D 1193) Nước thuốc thử – Yêu cầu kỹ thuật.
TCVN 2695 (ASTM D 974) Sản phẩm dầu mỏ – Xác định trị số axit và kiềm – Phương pháp chuẩn độ với chỉ thị màu.
TCVN 6777 (ASTM D 4057) Dầu mỏ và sản phẩm dầu mỏ – Phương pháp lấy mẫu thủ công.
ASTM D 3339 Test method for acid number of petroleum products by semi-micro color-indicator titration (Phương pháp xác định trị số axit của các sản phẩm dầu mỏ bằng chuẩn độ bán vi lượng theo chỉ thị mầu).
ASTM D 4177 Practice for Automatic Sampling of Petroleum and Petroleum Products (Dầu mỏ và sản phẩm dầu mỏ – Phương pháp lấy mẫu tự động).
ASTM E 177 Practice for use of the terms precision and bias in ASTM test methods – Hướng dẫn sử dụng các thuật ngữ độ chụm và độ chệch trong phương pháp thử ASTM.
3. Thuật ngữ và định nghĩa
3.1. Định nghĩa các thuật ngữ
3.1.1. Trị số axit (acid number)
Lượng bazơ quy định, tính bằng miligam kali hydroxit trên một gam mẫu, cần để chuẩn độ mẫu trong dung môi xác định đến điểm cuối xác định bằng cách sử dụng hệ thống phát hiện quy định.
3.1.1.1. Giải thích: Phương pháp này biểu thị lượng bazơ tính bằng số miligam kali hydroxit trên một gam mẫu cần để chuẩn độ một mẫu trong hỗn hợp của toluen và propan-2-ol có cho thêm một ít nước, từ số đọc ban đầu tới số đọc tính bằng milivôn đến số đọc tính bằng milivon tương ứng với dung dịch đệm kiềm trong nước, hoặc tới điểm uốn được xác định rõ theo quy định của phương pháp này.
3.1.1.2. Giải thích: Phương pháp này cung cấp các thông tin bổ sung. Lượng bazơ, tính bằng miligam kali hydroxit cần để chuẩn độ một gam mẫu trong dung môi từ số đọc ban đầu bằng milivon tới số đọc bằng milivon, tương ứng với dung dịch đệm axit trong nước mới được chuẩn bị, hoặc tới điểm uốn được xác định rõ theo quy định trong phương pháp thử này phải được báo cáo là trị số axit mạnh.
3.1.1.3. Giải thích: Nguyên nhân và ảnh hưởng của axit mạnh và các axit khác có thể rất khác nhau. Vì vậy, người sử dụng phương pháp thử này phải phân biệt và báo cáo cả hai khi phát hiện.
4. Tóm tắt phương pháp
4.1. Mẫu được hòa tan trong dung môi chuẩn độ và được chuẩn độ điện thế với dung dịch kali hydroxit trong alcoho, sử dụng điện cực chỉ thị thủy tinh và điện cực so sánh hoặc điện cực kết hợp. Các số liệu hiển thị trên máy được vẽ bằng tay hoặc ghi tự động theo các thể tích dung dịch chuẩn độ tương ứng. Các điểm cuối chỉ được xác định bằng các điểm uốn rõ ràng trên đường cong chuẩn độ. Nếu các điểm uốn không rõ ràng đối với dầu đã sử dụng, thì các điểm cuối được lấy theo số đọc trên máy tương ứng khi dùng các dung dịch đệm kiềm và axit trong nước.
5. Ý nghĩa và ứng dụng
5.1. Các sản phẩm dầu mỏ mới và đã sử dụng, điêzen sinh học và hỗn hợp điêzen sinh học có thể chứa axit có trong các phụ gia hoặc trong các sản phẩm sinh ra trong quá trình biến chất của dầu khi sử dụng như các sản phẩm oxy hóa. Lượng axit của các chất như vậy có thể xác định bằng chuẩn độ kiềm. Trị số axit là số đo lượng axit trong dầu dưới những điều kiện của phép thử. Trị số axit được dùng như hướng dẫn trong việc kiểm tra chất lượng khi pha chế dầu bôi trơn. Đôi khi nó được dùng như số đo mức độ giảm chất lượng của dầu bôi trơn trong quá trình sử dụng. Giới hạn để loại bỏ dầu phải được thiết lập theo kinh nghiệm.
5.2. Do sự đa dạng của các sản phẩm oxy hóa làm ảnh hưởng tới trị số axit và các axit hữu cơ lại rất khác nhau về tính ăn mòn, cho nên phương pháp này không được dùng để phán đoán tính ăn mòn của dầu hoặc điêzen sinh học và hỗn hợp trong điều kiện sử dụng. Không có mối liên hệ chung nào giữa trị số axit và xu hướng ăn mòn của điêzen sinh học và các hỗn hợp hoặc của dầu đối với kim loại.
6. Thiết bị, dụng cụ
6.1. Thiết bị chuẩn độ bằng tay
6.1.1. Vôn kế hoặc máy đo điện thế, có độ chính xác ± 0,005 V và độ nhạy ± 0,002 V, trên toàn dải đo ít nhất là ± 0,5 V, khi đồng hồ đo được dùng với các điện cực quy định trong 6.1.2 và 6.1.3 và khi điện trở giữa các điện cực nằm trong khoảng từ 0,2 MW đến 20 MW. Đồng hồ phải được bảo vệ tránh khỏi trường tĩnh điện rải rác bằng dây nối đất, sao cho không có sự thay đổi thường xuyên về giá trị đo trong toàn vùng đo sinh ra, do chạm tay vào mặt ngoài của điện cực thủy tinh, dây dẫn của điện cực thủy tinh, giá chuẩn độ hoặc chạm vào đồng hồ.
CHÚ THÍCH 4: Thiết bị thích hợp bao gồm một vôn kế điện tử có số đọc–liên tục được thiết kế để làm việc với dòng điện vào nhỏ hơn 5 x 10-12 A, khi hệ thống điện cực có điện trở 1 000 MW được nối qua cổng máy và có tấm chắn kim loại nối với đất, cũng như dây nối bảo vệ từ điện cực thủy tinh đến máy không bị nhiễu bởi trường tĩnh điện bên ngoài.
6.1.2. Điện cực cảm biến (sensing), có pH tiêu chuẩn, phù hợp với chuẩn độ không nước.
6.1.3. Điện cực so sánh, dùng điện cực so sánh bạc/chloride bạc (Ag/AgCI), nạp bằng LiCI 1M- 3M trong ethanol.
6.1.3.1. Điện cực kết hợp – Điện cực cảm biến có thể có điện cực so sánh Ag/AgCI gắn cùng trên ống của điện cực này, tạo thuận lợi làm việc chỉ với một điện cực. Điện cực kết hợp có ống nối bọc ngoài trên khoang so sánh và sử dụng chất điện ly trơ là ethanol, ví dụ LiCl 1M-3M trong ethanol. Các điện cực kết hợp này có cùng độ nhạy hoặc độ nhạy tốt hơn so với hệ thống điện cực đôi. Điện cực này có các tay áo tháo lắp được, dễ súc rửa và cho các chất điện ly vào.
CHÚ THÍCH 5: Điện cực thứ 3, như điện cực platin có thể được sử dụng để tăng độ ổn định của điện cực trong hệ thống.
6.1.4. Máy khuấy cơ học có tốc độ thay đổi, loại thích hợp có trang bị cánh khuấy loại cánh quạt. Tốc độ khuấy phải đủ mạnh nhưng không làm dung dịch bị bắn ra và cũng không để không khí lọt vào dung dịch. Cánh khuấy có bán kính 6 mm đặt nghiêng từ 30° đến 45°. Có thể dùng máy khuấy từ.
6.1.4.1. Nếu dùng máy khuấy điện thì nguồn điện phải phù hợp và tiếp đất sao cho khi tắt hoặc mở môtơ không làm biến đổi đáng kể số đo của vôn kế trong quá trình chuẩn độ.
6.1.5. Buret, dung tích 10 mL, có vạch chia 0,05 mL và đã hiệu chỉnh với độ chính xác ± 0,02 mL. Buret có đầu vuốt dài từ 100 mm đến 130 mm kể từ khóa và có khả năng đưa trực tiếp chất chuẩn độ vào bình chuẩn độ mà không bị tiếp xúc với không khí hoặc hơi xung quanh. Buret cho KOH phải có ống bảo vệ chứa vôi soda hoặc chất hấp thụ CO2 khác.
6.1.6. Cốc chuẩn độ, dung tích 250 mL làm từ thủy tinh borosilicat hoặc các vật liệu phù hợp khác.
6.1.7. Giá chuẩn độ, loại thích hợp để giữ các điện cực, que khuấy và buret.
CHÚ THÍCH 6: Sắp đặt sao cho việc di chuyển cốc chuẩn độ không ảnh hưởng tới điện cực, buret, máy khuấy.
6.2. Thiết bị chuẩn độ tự động
6.2.1. Các hệ thống chuẩn độ tự động phải có khả năng thực hiện được các phân tích cần thiết như quy định trong phương pháp này. Nói chung, hệ thống chuẩn độ tự động phải phù hợp với những đặc tính kỹ thuật như nêu ở 6.1.
6.2.2. Phải áp dụng chế độ bổ sung chất chuẩn liên tục. Trong quá trình chuẩn độ, tốc độ và thể tích chất chuẩn cho vào sẽ khác nhau, phụ thuộc vào tốc độ thay đổi của hệ thống. Khuyến cáo lượng thể tích thêm lớn nhất là 0,5 mL, và lượng thể tích thêm nhỏ nhất là 0,05 mL.
6.2.3. Ống đong có vạch chia, dung tích 50 mL, hoặc dụng cụ có sức chứa 50 mL ± 0,5 mL
6.2.4. Pipét, dung tích 2 mL, loại A
6.2.5. Cốc chuẩn độ, dung tích 250 mL, 125 mL hoặc có sức chứa phù hợp, làm từ thủy tinh dorosilicat hoặc các vật liệu phù hợp khác.
7. Thuốc thử và vật liệu
7.1. Độ tinh khiết của các thuốc thử – Trong toàn bộ các phép thử đều sử dụng các hóa chất cấp tinh khiết phân tích. Nếu không có quy định khác thì tất cả các thuốc thử phải phù hợp với các tiêu chuẩn hiện hành. Có thể sử dụng các loại khác, với điều kiện đảm bảo rằng các thuốc thử này có độ tinh khiết cao phù hợp, khi sử dụng không làm giảm độ chính xác của phép thử.
7.1.1. Có thể sử dụng các dung dịch bán sẵn trên thị trường thay cho việc phải chuẩn bị trong phòng thử nghiệm, với điều kiện là các dung dịch này phải được chứng nhận là tương đương.
7.1.2. Có thể chuẩn bị các thể tích khác của dung dịch, nhưng phải đảm bảo là nồng độ cuối cùng tương đương.
7.2. Độ tinh khiết của nước – Nếu không có quy định khác thì nước dùng trong thử nghiệm được hiểu là nước cất loại I, II hoặc III quy định trong TCVN 2117 (ASTM D 1193); hoặc loại nước cất có độ tinh khiết tương đương.
7.3. Chuẩn đầu – các mẫu thử theo quy định hoặc các mẫu thương phẩm có sẵn chuẩn đầu, được sử dụng tiêu chuẩn hóa các dung dịch chuẩn độ.
7.4. Ethanol, (Cảnh báo – Dễ cháy, độc, đặc biệt khi đã biến tính).
7.5. Liti chloride, LiCI.
7.6. Chất điện ly liti chloride, chuẩn bị dung dịch liti chloride 1 M đến 3 M trong ethanol.
7.7. Kali hydroxit, (Cảnh báo – Gây bỏng nghiêm trọng).
7.8. Propan-2-ol, khan (nhỏ hơn 0,1 % nước) (Cảnh báo – Dễ cháy). Nếu không có rượu khan thì có thể làm khan rượu bằng cách chưng cất với cột có nhiều đĩa. Loại bỏ 5 % phần cất đầu và dùng 95 % còn lại. Cũng có thể làm khan bằng cách cho chảy qua cột rây phân tử như loại rây kiểu Linde 4A, cho dung môi chảy qua cột rây phân tử, dùng một phần rây phân tử với 10 phần dung môi.
CHÚ THÍCH 7: Nếu không có chất ức chế từ đầu thì propan-2-ol có thể chứa peroxit. Khi đó có thể gây nổ, nếu bảo quản trong bình chứa hoặc các dụng cụ khác như chai chỉ còn ít hoặc chai đã gần hết.
7.9. Dung dịch đệm nước pH 4, pH 7 và pH 11 – Các dung dịch này được thay thường xuyên để đảm bảo độ ổn định hoặc khi phát hiện sự nhiễm bẩn. Các thông tin về độ ổn định do nhà sản xuất cung cấp.
8. Hệ điện cực
8.1. Chuẩn bị các điện cực
Khi sử dụng điện cực so sánh Ag/AgCI để chuẩn độ, nếu chất điện ly không phải là LiCI 1 M – 3 M trong ethanol thì phải thay chất điện ly này. Tháo chất điện ly ở điện cực, rửa sạch các muối (nếu có) bằng nước, sau đó bằng ethanol. Tráng vài lần bằng dung dịch điện ly LiCI. Cuối cùng là thay ống nối có bọc ngoài và nạp chất điện ly LiCI vào điện cực cho đến tận nút xả. Khi nạp, ống nối có bọc ngoài phải đảm bảo không còn chất điện ly chảy vào hệ thống. Điện cực kết hợp cũng được chuẩn bị theo cách tương tự. Dùng dụng cụ hút chân không để loại bỏ chất điện ly trong điện cực kết hợp.
8.2. Kiểm tra các điện cực
Kiểm tra tổ hợp máy đo – điện cực khi đưa vào sử dụng lần đầu, khi lắp đặt các điện cực mới và khi kiểm tra dụng cụ theo định kỳ. Rửa các điện cực bằng dung môi, sau đó bằng nước. Nhúng các điện cực vào dung dịch đệm nước pH 4, đọc giá trị mV sau khi khuấy trong 1 min. Lấy điện cực ra và rửa bằng nước. Nhúng các điện cực vào dung dịch đệm nước pH 7. Đọc giá trị mV sau khi khuấy trong 1 min. Hệ thống điện cực tốt nếu có sự chênh lệch ít nhất bằng 162 mV (20 °C đến 25 °C). Nếu chênh lệch nhỏ hơn 162 mV, nâng ống nối của điện cực lên và đảm bảo dòng chất điện ly liên tục. Lặp lại phép đo. Nếu chênh lệch vẫn nhỏ hơn 162 mV, cần làm sạch hoặc thay (các) điện cực.
8.2.1. Khi hai điện cực cảm biến và điện cực so sánh tách riêng, một đôi điện cực được coi là một bộ. Nếu thay một trong hai điện cực thì sẽ coi như là một đôi khác và lúc đó phải kiểm tra lại.
8.3. Bảo dưỡng và bảo quản các điện cực
Rửa cẩn thận điện cực, giữ khớp nối nhám không bị các chất bẩn lạ bám vào. Cần kiểm tra điện cực thường xuyên để thu được kết quả đo thế lặp lại, vì các chất bẩn có thể tạo ra thế tiếp xúc với chất lỏng không ổn định và không dễ nhận biết. Điều này sẽ không quan trọng nếu điểm cuối định phân được chọn là điểm uốn của đường cong chuẩn độ, hoặc cũng có thể rất cần thiết nếu điểm cuối được chọn là một thế tự quy định.
CHÚ THÍCH 8: Xem Phụ lục A về quy trình kiểm tra tính năng của điện cực.
8.3.1. Chu kỳ làm sạch điện cực thủy tinh tùy thuộc vào thời gian sử dụng và loại các mẫu phân tích (ít nhất mỗi tuần một lần khi dùng liên tục) bằng cách nhúng trong dung dịch làm sạch oxy hóa mạnh không chứa crom. Điện cực so sánh được làm sạch định kỳ khi sử dụng và khi thay điện cực mới. Ít nhất mỗi tuần một lần, tháo khô điện cực so sánh, sau đó nạp lại chất điện ly LiCI mới cho đến tận nút xả. Phải đảm bảo sao cho không có bọt khí lọt vào chất lỏng của điện cực. Nếu quan sát thấy có các bọt khí thì giữ thẳng đứng điện cực và gõ nhẹ để đuổi các bọt khí ra. Luôn duy trì chất điện ly trong điện cực so sánh trên mức chất lỏng trong cốc chuẩn độ hoặc trong bình.
8.3.2. Trước mỗi lần chuẩn độ, nhúng các điện cực trong nước (pH từ 4,5 đến 5,5) trong 5 min. Ngay trước khi sử dụng phải rửa các điện cực bằng propan-2-ol, sau đó bằng dung môi chuẩn độ.
8.3.3. Khi không sử dụng, ngâm nửa dưới của điện cực so sánh trong chất điện ly LiCI. Khi sử dụng điện cực thủy tinh, bảo quản điện cực này trong nước đã được axit hóa với HCI đến pH từ 4,5 đến 5,5. Không cho phép nhúng các điện cực trong dung môi chuẩn độ trong thời gian không chuẩn độ. Vì các điện cực mỏng, dễ vỡ, phải thận trọng khi sử dụng.
8.3.3.1. Tuổi thọ của điện cực – Tùy theo mục đích sử dụng, thường dùng các điện cực từ 3 tháng đến 6 tháng. Các điện cực có giới hạn sử dụng riêng, trước khi dùng cần kiểm tra (Xem 8.2).
9. Chuẩn hóa dụng cụ
9.1. Xác định số đọc trên máy đối với các dung dịch đệm nước – Để đảm bảo sự lựa chọn một cách tương đối các điểm cuối khi không nhận được những điểm uốn rõ ràng trên các đường cong chuẩn độ, thì người ta xác định hàng ngày những giá trị đọc trên máy cho mỗi cặp điện cực trên các dung dịch đệm axit hoặc kiềm trong nước.
CHÚ THÍCH 9: Sự phản ứng của các điện cực thủy tinh khác nhau đối với hoạt tính ion hydro không giống nhau. Vì vậy, cần định kỳ xác định giá trị đọc trên máy cho từng hệ điện cực, phù hợp với các dung dịch đệm đã được lựa chọn theo quy ước để tìm điểm cuối axit hoặc kiềm.
9.2. Nhúng các điện cực trong dung dịch đệm có pH 4 và pH 11, và khuấy từng loại trong khoảng 5min, duy trì nhiệt độ của dung dịch đệm tại nhiệt độ chuẩn độ chính xác đến ± 2 °C. Đọc thế trên đồng hồ. Giá trị đọc được coi là điểm cuối trên đường cong chuẩn độ khi không xác định được điểm uốn.
10. Chuẩn bị mẫu dầu đã sử dụng
10.1. Trong trường hợp này, tuân thủ chặt chẽ quy trình lấy mẫu là cần thiết, vì các cặn chính là axit hoặc kiềm hoặc cặn đã hấp thụ axit hoặc kiềm có trong mẫu. Khi lấy mẫu đại diện, nếu có sai sót sẽ gây ra những sai lỗi nghiêm trọng.
10.1.1. Tùy từng trường hợp, áp dụng kỹ thuật lấy mẫu theo TCVN 6777 (ASTM D 4057) (Lấy mẫu thủ công) hoặc ASTM D 4177 (Lấy mẫu tự động).
10.1.2. Khi lấy mẫu các dầu bôi trơn đã qua sử dụng, mẫu phải đảm bảo đại diện và không bị nhiễm bẩn từ bên ngoài vào.
CHÚ THÍCH 10: Do dầu đã sử dụng có thể thay đổi trong khi tồn chứa, cho nên cần phân tích càng sớm càng tốt các mẫu sau khi lấy khỏi hệ thống bôi trơn. Ghi ngày tháng lấy mẫu và ngày tháng phân tích.
10.2. Đun nóng mẫu dầu (xem Chú thích 11) đã sử dụng trong vật chứa ban đầu lên 60 °C ± 5 °C rồi khuấy cho đến khi cặn lơ lửng đều trong dầu. Nếu vật chứa ban đầu là can hoặc cốc chứa đầy quá 3/4 dung tích, chuyển toàn bộ mẫu vào một chai thủy tinh không màu có dung tích lớn hơn 1/3 thể tích mẫu. Chuyển tất cả cặn từ vật chứa ban đầu vào chai này bằng cách khuấy mạnh nhiều lần từng lượng nhỏ mẫu trong vật chứa ban đầu.
CHÚ THÍCH 11: Khi thấy mẫu không có cặn thì có thể không cần đun nóng.
10.3. Khi tất cả các cặn đã lơ lửng hoàn toàn thì lọc mẫu qua lưới lọc loại 100 mesh để loại những hạt bẩn lớn.
CHÚ THÍCH 12: Khi thấy mẫu không có cặn lắng thì không cần lọc.
PHƯƠNG PHÁP THỬ A
11. Thuốc thử
11.1. Xem Điều 7.
11.2. Axit chlohydric (HCI) – Tỷ trọng tương đối 1,19 (Cảnh báo – Ăn mòn, gây bỏng).
11.3. Toluen, (Cảnh báo – Dễ cháy).
11.4. Dung dịch chuẩn axit chlohydric trong alcohol (0,1 mol/L). (Cảnh báo – Xem 11.2 và 7.8). Trộn 9 mL dung dịch axit HCI (tỷ trọng tương đối HCI là 1,19) với 1 L propan-2-ol khan. Chuẩn hóa thường xuyên để phát hiện những sự thay đổi độ chuẩn từ 0,0005 bằng chuẩn độ điện thế với khoảng 8 mL (đo chính xác) dung dịch KOH 0,1 mol/L trong alcohol đã được pha loãng với 125 mL nước không có CO2.
11.5. Dung dịch chuẩn kali hydroxit trong alcohol (0,1 mol/L). (Cảnh báo – Xem 7.7 và 7.8). Thêm 6 g KOH vào khoảng 1 L propan-2-ol khan. Đun sôi nhẹ trong 10 min để tác động dung dịch. Để yên dung dịch trong hai ngày, sau đó, lọc phần chất lỏng phía trên qua phễu thủy tinh thiêu kết mịn. Bảo quản dung dịch trong chai chịu hóa chất. Để tránh cho dung dịch khỏi sự xâm nhập của CO2 có trong không khí, người ra nối chai với ống chứa vôi soda hoặc chất hấp thụ silicat không-sợi. Không cho dung dịch tiếp xúc với nút lie, cao su, mỡ bôi nút nhám gốc xà phòng. Chuẩn hóa thường xuyên để phát hiện sự thay đổi độ chuẩn từ 0,0005 bằng chuẩn độ điện thế với một lượng cân kali axit phtalat hòa tan trong nước không chứa CO2.
11.6. Dung môi chuẩn độ
Cho 5 mL ± 0,2 mL nước vào 495 mL ± 5 mL propan-2-ol khan và trộn đều. Cho thêm 500 mL ± 5 mL toluen (Cảnh báo – Dễ cháy). Nên chuẩn bị dung môi chuẩn độ này với những lượng lớn và xác định mẫu trắng hàng ngày bằng chuẩn độ trước khi dùng.
11.7. Chloroform, (Cảnh báo – Dễ cháy. Là chất độc hại).
12. Xác định trị số axit và trị số axit mạnh
12.1. Cân lượng mẫu như đã hướng dẫn ở Bảng 1 (xem Chú thích 12), cho vào cốc dung tích 250 mL hoặc bình chuẩn độ phù hợp, rồi thêm vào 125 mL dung môi chuẩn độ (xem Chú thích 13). Chuẩn bị các điện cực theo quy định ở 8.2. Đặt cốc hoặc bình chuẩn độ vào giá, điều chỉnh sao cho các điện cực ngập khoảng một nửa. Mở máy khuấy, khuấy liên tục trong lúc chuẩn độ với tốc độ đủ mạnh nhưng không gây bắn tóe và không kéo theo không khí vào trong dung dịch.
Bảng 1 – Lượng mẫu khuyến cáo
|
Trị số axit |
Khối lượng mẫu, g |
Độ chính xác khi cân, g |
|
từ 0,05 đến 1,0 |
20,0 ± 2,0 |
0,10 |
|
từ 1,0 đến <> |
5,0 ± 0,5 |
0,02 |
|
từ 5 đến <> |
1,0 ± 0,1 |
0,005 |
|
từ 20 đến <> |
0,25 ± 0,02 |
0,001 |
|
từ 100 đến <> |
0,1 ± 0,01 |
0,0005 |
CHÚ THÍCH 13: Nếu nghi ngờ lượng mẫu như khuyến cáo gây cản trở các điện cực thì có thể lấy lượng mẫu nhỏ hơn. Các kết quả thử đối với các lượng mẫu nhỏ hơn có thể không tương đương với kết quả của lượng mẫu đã khuyến cáo. Quy định độ chụm không áp dụng cho các kết quả khi phân tích lượng mẫu nhỏ hơn.
CHÚ THÍCH 14: Dung môi chuẩn độ có chứa chloroform (Cảnh báo – Chloroform có thể gây chết người nếu hút phải. Độc nếu hít phải. Khi cháy sinh ra hơi độc) có thể dùng thay cho tôluen để hòa tan hoàn toàn những cặn nặng nào đó của các chất asphal. Khi dùng chloroform, các kết quả thử có thể không tương đương với các kết quả thử như dùng toluen. Không áp dụng quy định về độ chụm cho các kết quả khi dùng chloroform.
12.2. Dùng buret thích hợp, cho dung dịch KOH 0,1 mol/L vào trong buret rồi đặt buret vào thiết bị chuẩn độ, chú ý đặt buret sao cho đầu buret ngập sâu 25 mm trong chất lỏng của bình chuẩn độ. Ghi số đọc ban đầu của buret và thế trên máy.
12.3. Phương pháp chuẩn độ bằng tay
12.3.1. Thêm từng lượng nhỏ dung dịch rượu KOH 0,1 mol/L rồi đợi cho đến khi đạt thế cân bằng. Ghi số đọc của buret và số đọc trên máy.
12.3.2. Lúc bắt đầu chuẩn độ và ở vài vùng uốn tiếp theo khi thêm 0,1 mL dung dịch KOH 0,1 mol/L thường có sự thay đổi lớn hơn 30 mV, thì chỉ thêm từng lượng 0,05 mL dung dịch KOH.
12.3.3. Những đoạn trung gian (phẳng) khi thêm 0,1 mL dung dịch rượu KOH 0,1 mol/L mà thay đổi ít hơn 30 mV thì thêm nhiều hơn sao cho sự thay đổi thế xấp xỉ bằng nhau, nhưng không lớn hơn 30 mV.
12.3.4. Chuẩn độ theo cách trên cho đến khi thế thay đổi nhỏ hơn 5 mV/0,1 mL KOH và cho thấy dung dịch có độ kiềm hơn dung dịch đệm kiềm nước.
12.3.5. Chuyển dung dịch chuẩn độ khỏi hệ chuẩn độ. Dùng dung môi chuẩn độ tráng điện cực và đầu buret sau đó tráng bằng rượu propan-2-ol, rồi tráng bằng nước cất. Nhúng điện cực vào trong nước cất ít nhất 5 min trước khi dùng cho lần chuẩn độ khác để hoàn lại lớp gel nước của điện cực thủy tinh. Sau 5 min ngâm trong nước, tráng bằng propan-2-ol và dung môi chuẩn độ trước khi chuyển sang lần chuẩn độ tiếp theo. Nếu điện cực bị phát hiện là bẩn hay bị nhiễm bẩn, xử lý như nêu ở 8.1. Bảo quản các điện cực theo 8.3.3.
12.4. Phương pháp chuẩn độ tự động
12.4.1. Điều chỉnh dụng cụ theo hướng dẫn của nhà sản xuất cho phù hợp những yêu cầu về cân bằng thế khi thêm chất chuẩn độ.
12.4.2. Để đảm bảo thiết bị xác định lượng axit mạnh chỉ ra sự có mặt của các axit này, cần kiểm tra số mV ban đầu của mẫu thử tương ứng với số đọc mV của dung dịch đệm axit trong nước. Ghi lại thể tích của KOH đã cho vào để đạt giá trị mV của dung dịch đệm nước pH 4. Giá trị này dùng để tính trị số axit mạnh. Thực hiện theo quy trình chuẩn độ tự động và ghi đường cong thế hoặc đường cong vi phân, tùy từng trường hợp.
12.4.3. Chuẩn độ bằng dung dịch KOH 0,1 mol/L. Điều chỉnh thiết bị hoặc chương trình sao cho thu được điểm uốn phù hợp để tính kết quả, tốc độ và thể tích chất chuẩn độ cho vào là cơ sở của sự thay đổi độ dốc của đường cong chuẩn độ. Cho chất chuẩn độ vào từng lượng phù hợp để đạt chênh lệch thế từ 5 mV đến 15 mV trên một lần. Thể tích lượng chất chuẩn độ thêm vào mỗi lần nằm trong khoảng 0,05 mL và 0,5 mL. Cho chất chuẩn độ tiếp nếu có tín hiệu không thay đổi trên 10 mV trong 10 s. Thời gian đợi nhiều nhất giữa các lần không quá 60 s.
12.4.4. Kết thúc chuẩn độ khi tín hiệu chất đệm đạt 11 pH, điện thế vượt qua 200 mV. Điểm tương đương có thể nhận biết được khi vi phân bậc 1 của đường cong chuẩn độ là cực đại, điểm này cao hơn hẳn sự nhiễu sinh ra do các ảnh hưởng của tĩnh điện. Xem thêm 13.1.1.
12.4.5. Rửa sạch cặn dư bám trên mẫu lần trước và hydrat hóa lại điện cực. Khi chuẩn độ xong, tráng các điện cực và đầu buret bằng dung môi chuẩn độ. Nếu sạch, nhúng tiếp vào propan-2-ol, sau đó là nước. Nhúng các điện cực vào trong nước có pH 4,5 – 5,5 ít nhất 3 min – 5 min để hydrat hóa lại lớp gel nước của điện cực thủy tinh. Nhúng vào propan-2-ol để loại bỏ nước trước khi bắt đầu thử mẫu tiếp theo. Nếu mẫu vẫn bám cặn sau khi rửa trong dung môi chuẩn độ, có thể rửa bằng cách thêm dung môi như toluen, xilen, heptan, hoặc chloroform. Nếu cốc chứa dung môi được khuấy mạnh, sẽ đạt được hiệu quả cao khi rửa. Khi sử dụng thiết bị tự động, làm sạch bằng cách rửa với dung môi chuẩn độ, sau đó vừa ngâm vừa khuấy trong dung môi như toluen, xylen, heptan hoặc chloroform trong thời gian 45 s, rồi ngâm trong propan-2-ol một thời gian ngắn để loại bỏ dung môi, sau đó ngâm trong nước có pH 4,5 – 5,5 từ 3 min – 5 min để hydrat hóa lại. Ngâm trong propan-2-ol một thời gian ngắn để loại bỏ nước trước khi thử mẫu tiếp theo. Cốc chứa dung môi làm sạch, cốc chứa propan-2-ol và cốc chứa nước giống nhau để có thể dùng cho một loạt thử ngắn của mẫu. Cốc phải được thay trong khoảng thời gian hợp lý, trước khi có sự tích lũy cặn bẩn. Người dùng phải đảm bảo rằng điện cực sạch và đã hydrat hóa. Nếu thấy các điện cực bị bẩn thì xử lý như 8.1. Bảo quản điện cực theo 8.3.3
CHÚ THÍCH 15: Khi dự kiến trị số axit bằng hoặc nhỏ hơn 0,1, có thể nhận được độ chụm tốt hơn khi thay đổi phương pháp theo một trong các cách sau, thay bằng dung dịch 0,01 hoặc 0,05 M KOH; tăng lượng mẫu lên 20 g hoặc chuyển từ việc dùng buret thủ công (có chia 0,05 mL) sang buret tự động, buret này có thể truyền các lượng dung dịch KOH nhỏ hơn, nếu các mẫu đang được phân tích theo phương pháp chuẩn độ bằng tay.
12.5. Mẫu trắng
12.5.1. Đối với từng bộ mẫu và từng đợt dung môi chuẩn độ mới, chuẩn bị mẫu trắng bằng 125 mL dung môi. Đối với chuẩn độ bằng tay, thêm từng lượng từ 0,01 mL đến 0,05 mL dung dịch KOH 0,1 mol/L, đợi cho thế ổn định rồi mới cho thêm những lượng tiếp theo. Ghi các số đọc của buret và máy khi đạt cân bằng sau mỗi lần thêm. Đối với chuẩn độ tự động tiến hành theo cùng phương pháp chuẩn độ như xác định tính axit của mẫu, nhưng sử dụng các lượng chất chuẩn độ nhỏ hơn từ 0,01 mL đến 0,05 mL. Định kỳ kiểm tra lại mẫu trắng trên cơ sở khối lượng mẫu.
12.5.2. Khi có axit mạnh và cần xác định trị số axit mạnh, chuẩn bị mẫu trắng bằng 125 mL dung môi chuẩn độ, thêm từng lượng từ 0,01 mL đến 0,05 mL dung dịch rượu HCI 0,1 mol/L theo như cách tương tự quy định ở 12.5.1.
13. Tính kết quả
13.1. Đối với chuẩn độ bằng tay
Vẽ đồ thị thể tích thêm vào của dung dịch KOH 0,1 mol/L theo giá trị đọc được của máy (xem Hình 1). Chỉ coi là điểm cuối khi thấy điểm uốn rõ (Chú thích 16), có giá trị sát nhất với giá trị điện thế xác định bằng dung dịch đệm axit hoặc kiềm trong nước. Nếu không có điểm uốn hoặc khó nhận thấy, (xem Hình 1, đường cong B) thì lấy điểm cuối là giá trị thu được trên máy nhờ các dung dịch đệm nước thích hợp.
CHÚ THÍCH 16: Nói chung một điểm uốn được nhận ra khi kiểm tra thấy vài lượng 0,05 mL dung dịch chuẩn độ thêm vào gây ra sự thay đổi về thế trên 15 mV, tức là lớn hơn ít nhất 30% so với sự thay đổi có được khi thêm những lượng như vậy ở trước và sau điểm đó. Nói chung các điểm uốn xác định chỉ có thể nhận biết được trong những vùng khảo sát với những lượng thêm như nhau.
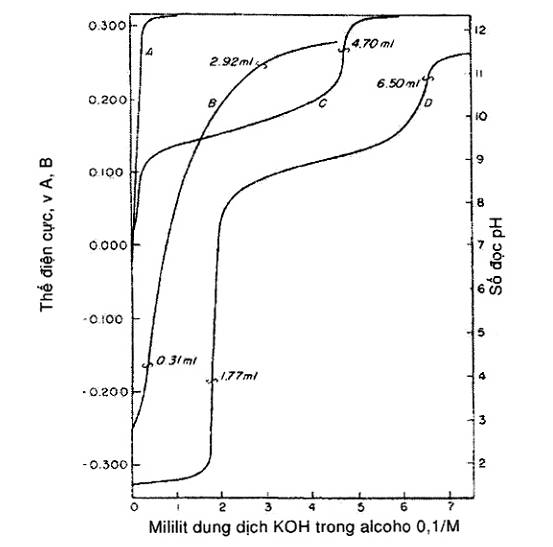
CHÚ DẪN
Đường cong A – Mẫu trắng cho 125 mL dung môi chuẩn độ
Đường cong B – 10,00 g dầu trục khuỷu đã sử dụng cộng 125 mL dung môi chuẩn độ. Do điểm uốn không rõ, chọn các điểm cuối ở các số đọc của máy khi tiến hành theo hai dung dịch đệm nước.
Đường cong C – 10,00 g dầu có chứa axit yếu và mạnh cộng 125 mL dung môi chuẩn độ. Chọn điểm cuối tại vị trí mà ở đó đường cong gần như thẳng đứng.
Đường cong D – 10,00 g dầu có chứa axit yếu cộng 125 mL dung môi chuẩn độ. Chọn các điểm cuối tại vị trí mà ở đó đường cong gần như thẳng đứng.
Hình 1 – Các đường cong chuẩn độ
13.1.1. Một vài loại phụ gia hóa học có thể cho điểm uốn theo điểm cuối của dung dịch đệm. Đối với các phụ gia thì lấy điểm uốn cuối cùng để tính toán. Nếu dùng máy chuẩn độ tự động, có thể phải thay đổi các thông số của thiết bị để phát hiện loại điểm cuối này.
13.1.2. Đối với mọi chuẩn độ axit cho các dầu đã sử dụng thì điểm cuối là điểm trên đường cong chuẩn độ, tương ứng với số đọc của máy được xác định bằng dung dịch đệm kiềm nước (pH 11), và số đọc của máy được xác định bằng dung dịch đệm axit nước (pH 4), chỉ ra sự có mặt các axit mạnh.
CHÚ THÍCH 17: Theo các nghiên cứu hợp tác để xác định trị số axit trên các dầu mới, dầu đã sử dụng và các phụ gia đậm đặc cho thấy đối với dầu mới và phụ gia đậm đặc thì dễ xác định được điểm uốn còn đối với các dầu đã sử dụng, nói chung khó xác định được điểm uốn hoặc không thấy có điểm uốn.
13.2. Phương pháp chuẩn độ tự động
Đánh dấu điểm cuối trên đường cong thu được tại 11.4, giống như cách khi chuẩn độ bằng tay.
13.3. Phương pháp tính toán
Áp dụng phương pháp nêu ở 12.3.1 để tính chuẩn độ bằng tay và tự động.
13.3.1. Tính trị số axit và trị số axit mạnh như sau:
Trị số axit, mg KOH/g = (A – B) x M x 56,1/W (1)
Trị số axit mạnh, mg KOH/g = (CM – Dm) x 56,1/W (2)
trong đó
A là thể tích dung dịch KOH đã dùng để chuẩn độ mẫu tới điểm cuối, khi xuất hiện số đọc trên máy của điểm uốn sát nhất với số đọc trên máy của dung dịch đệm nước pH 11, hoặc nếu không có điểm uốn hoặc khó xác định, thì điểm cuối xác định được nhờ số đọc tương ứng trên máy của dung dịch đệm nước pH 11, tính bằng mililit. Đối với các phụ gia, A là thể tích của KOH tại điểm uốn cuối cùng;
B là thể tích tương ứng với A dùng để chuẩn độ mẫu trắng, tính bằng mililit;
M là nồng độ của dung dịch KOH, tính bằng mol trên lít;
M là nồng độ của dung dịch HCI, tính bằng mol trên lít;
W là khối lượng mẫu, tính bằng gam;
C là thể tích dung dịch KOH đã dùng để chuẩn mẫu tới điểm cuối, khi xuất hiện số đọc của máy tương ứng với dung dịch đệm nước pH 4, tính bằng mililit;
D là thể tích dung dịch HCI dùng để chuẩn độ mẫu trắng là dung môi tới điểm cuối tương ứng với C, tính bằng mililit.
14. Kiểm soát chất lượng
14.1. Kiểm tra xác nhận khả năng áp dụng của phương pháp bằng cách phân tích mẫu kiểm soát chất lượng (QC), mẫu này phải đại diện cho các mẫu được phân tích điển hình.
CHÚ THÍCH 18: Các dầu đã dùng đặc biệt là dầu động cơ bị thay đổi trong quá trình tồn chứa, do vậy các mẫu có thể không phù hợp cho mục đích này.
14.2. Trước khi thực hiện các phép đo, thí nghiệm viên cần xác định giá trị trung bình và các giới hạn kiểm soát của mẫu kiểm soát chất lượng (QC).
14.3. Ghi các kết quả QC và phân tích theo biểu đồ hoặc kỹ thuật thống kê tương đương để đảm bảo việc kiểm soát mang tính thống kê cho toàn bộ quá trình thử nghiệm. Các số liệu ngoài vùng kiểm soát phải được nghiên cứu kỹ để tìm các nguyên nhân chính. Các kết quả này có thể dùng khi hiệu chuẩn thiết bị, nhưng không nhất thiết.
14.4. Tần suất thử nghiệm QC phụ thuộc vào tầm quan trọng của chỉ tiêu chất lượng đang kiểm tra, độ ổn định của quy trình thử, và các yêu cầu của khách hàng. Thông thường, hàng ngày phân tích mẫu QC, nếu số lượng mẫu đem phân tích lớn thì tăng tần suất số phân tích mẫu kiểm soát chất lượng lên. Tuy nhiên, khi công bố là phép thử đang được tiến hành dưới điều kiện kiểm soát mang tính thống kê thì có thể giảm tần suất thử nghiệm. Độ chụm của mẫu kiểm soát chất lượng phải được kiểm tra định kỳ theo độ chụm quy định trong phần độ chụm và độ chệch của tiêu chuẩn để đảm bảo chất lượng các số liệu.
14.5. Khuyến cáo mẫu QC được phân tích thường xuyên là đại diện cho các mẫu phân tích hàng loạt. Nguồn cung cấp mẫu phải sẵn sàng đáp ứng cho thời gian sử dụng dự kiến, mẫu phải đảm bảo đồng nhất, ổn định dưới điều kiện bảo quản quy định. Trị số kiềm của mẫu có thể bị thay đổi dưới điều kiện bảo quản, khi không kiểm soát được tình hình thì độ ổn định của mẫu QC có thể chính là nguyên nhân gây sai số.
15. Báo cáo kết quả
15.1. Có hai cách khác nhau để xác định điểm cuối, báo cáo loại điểm cuối đã sử dụng là điểm uốn hoặc điểm cuối của chất đệm. Báo cáo lượng mẫu đã sử dụng nếu khác với lượng mẫu khuyến cáo. Đồng thời cũng phải báo cáo nếu sử dụng chloroform làm dung môi. Báo cáo kết quả trị số axit hoặc trị số axit mạnh như sau:
Trị số axit (TCVN 6325, Phương pháp thử A) = (kết quả) (3)
Trị số axit mạnh (TCVN 6325, Phương pháp thử A) = (kết quả) (4)
15.2. Đối với các mẫu dầu đã sử dụng, báo cáo ngày tiến hành thử nghiệm, ngày nhận mẫu và ngày lấy mẫu (xem 10.2).
16. Độ chụm và độ chệch
16.1. Trị số axit
16.1.1. Độ lặp lại – Sự chênh lệch giữa hai kết quả thử liên tiếp nhận được do cùng một thí nghiệm viên tiến hành trên cùng một thiết bị, dưới các điều kiện thử không đổi, trên cùng một mẫu thử, trong một thời gian dài với thao tác bình thường và chính xác của phương pháp thử này, chỉ một trong hai mươi trường hợp được vượt các giá trị sau:
Dầu mới = 0,044 (X + 1) (5)
Dầu đã sử dụng, điểm cuối của dung dịch đệm = 0,117X (6)
trong đó
X là giá trị trung bình của hai kết quả thử.
16.1.2. Độ tái lập – Sự chênh lệch giữa hai kết quả thử độc lập, nhận được do hai thí nghiệm viên khác nhau làm việc trong hai phòng thử nghiệm khác nhau, trên cùng một mẫu thử, trong một thời gian dài với thao tác bình thường và chính xác của phương pháp thử này, chỉ một trong hai mươi trường hợp được vượt các giá trị sau:
Dầu mới = 0,141 (X + 1) (7)
Dầu đã sử dụng, điểm cuối của dung dịch đệm = 0,44X (8)
trong đó
X là giá trị trung bình của hai kết quả thử.
16.2. Trị số axit mạnh
Số liệu về độ chụm của trị số axit mạnh chưa được nghiên cứu xây dựng vì cũng rất ít xuất hiện khi phân tích mẫu.
16.3. Độ chệch
Tiêu chuẩn này không quy định độ chệch, vì trị số axit chỉ được xác định theo phương pháp này.
PHƯƠNG PHÁP THỬ B
17. Thuốc thử và vật liệu
17.1. Xem Điều 7.
17.2. Dung dịch chuẩn kali hydroxit trong alcohol (0,01 mol/L). (Cảnh báo – xem 7.7 và 7.8). Thêm 0,56 g kali hydroxit (KOH) vào khoảng 1 L propan-2-ol hoặc cân 1,122 g ± 0,02 g dung dịch 50 % KOH cho vào 1 L propan-2-ol khan. Đun sôi nhẹ trong 10 min để tác động dung dịch. Để yên dung dịch, tránh carbon dioxit (CO2) trong 2 ngày, sau đó lọc phần chất lỏng phía trên qua phễu thủy tinh thiêu kết mịn. Bảo quản dung dịch trong chai chịu hóa chất. Để tránh cho dung dịch khỏi sự xâm nhập của CO2 có trong không khí, người ta nối chai với ống chứa vôi soda hoặc chất hấp thụ soda silicat không sợi. Không cho dung dịch tiếp xúc với nút lie, cao su, mỡ bôi nút nhám gốc xà phòng. Chuẩn hóa thường xuyên để phát hiện sự thay đổi nồng độ từ 0,0005 mol/L bằng chuẩn độ điện thế với một lượng pipet dung dịch kali axit phtalat hòa tan trong nước không chứa CO2.
17.3. Kali axit phtalat (KHC8H4O4), chuẩn đầu, khô – Lấy 10 g đến 20 g kali axit phtalat chuẩn đầu, sàng qua lưới cỡ 100 mesh, rồi cho vào trong bình cân ở 120 °C trong 2 h. Đậy nắp bình và làm nguội trong bình hút ẩm.
17.4. Dung dịch kali axit phtalat (KHP) (0,01 mol/L) – Đối với dung dịch chuẩn độ, cân khoảng 1g kali axit phtalat chuẩn đầu, khô (KHC8H4O4), chính xác đến ± 0,001 g, cho vào bình định mức dung tích 500 mL, dùng nước DI loại II pha loãng đến vạch mức. Ngoài ra, đối với việc chuẩn hóa trên cơ sở khối lượng, cân KHP, chính xác đến 0,0001g, và ghi lại tổng lượng nước và KHP chính xác đến ± 0,01 g, nồng độ của dung dịch được biểu thị theo mg KHP/g. Trộn kỹ dung dịch để hòa tan hoàn toàn.
18. Cách tiến hành
18.1. Chuẩn hóa chất chuẩn độ KOH 0,01 M trong alcohol
18.1.1. Cân 2 g dung dịch KHP rồi cho vào cốc dung tích 125 mL hoặc cốc có kích cỡ phù hợp và ghi lại khối lượng chính xác đến 0,0001 g nếu sử dụng phép tiêu chuẩn trên cơ sở khối lượng hoặc sử dụng pipet lấy 2 mL dung dịch KHP cho vào trong bình và thêm khoảng 50 mL nước không chứa CO2. Chuẩn độ dung dịch để xác định độ chuẩn của KOH 0,01 M. Thể tích này của dung dịch KHP sẽ dùng hết khoảng 2 mL KOH 0,01 M.
18.1.2. Chuẩn bị thêm hai dung dịch KHP để chuẩn hóa chất chuẩn độ như trong 18.1.
18.1.3. Dùng ba phép xác định để tính nồng độ trung bình (mol/L) của KOH. Giá trị trung bình xác định chất chuẩn độ mol/L được chấp nhận ± 0,0005 M.
18.2. Xác định mẫu dung môi trắng
Điều chỉnh thiết bị theo chỉ dẫn của nhà sản xuất để cung cấp phương thức chuẩn độ động học. Thêm dần thể tích chuẩn độ nhưng không được lớn hơn 0,2 mL. Lấy chính xác 50 mL propan-2-ol sử dụng xylanh chia độ, pipet, hoặc thiết bị lấy tự động có dung tích 50 mL ± 0,5 mL vào trong cốc dung tích 125 mL hoặc cốc có kích cỡ phù hợp. Khuấy dung dịch và chuẩn độ. Ghi lại thể tích KOH chính xác đến ± 0,01 mL dùng để chuẩn độ đến điểm uốn.
18.3. Phân tích mẫu
18.3.1. Điều chỉnh thiết bị theo chỉ dẫn của nhà sản xuất để cung cấp phương thức thêm dung dịch chuẩn độ.
18.3.2. Cân 5 g điêzen sinh học rồi cho vào bình dung tích 125 mL hoặc bình có kích cỡ phù hợp, trên cân phân tích và ghi lại khối lượng chính xác đến 0,0001 g. Lấy 50 mL ± 0,50 mL IPA sử dụng pipet hoặc thiết bị lấy tự động cho vào trong cốc phù hợp. Chuẩn bị các điện cực như trong 8.1. Đặt cốc hoặc bình chuẩn độ vào giá chuẩn độ, điều chỉnh sao cho các điện cực ngập khoảng một nửa. Khởi động máy khuấy, khuấy liên tục trong lúc chuẩn độ với tốc độ đủ mạnh nhưng không gây bắn tóe và không kéo theo không khí vào trong dung dịch.
CHÚ THÍCH 19: Điều quan trọng khi dùng cùng một thể tích dung môi chuẩn độ ± 0,5 mL dùng cho mẫu trắng và mẫu thử có thể cho các kết quả khác nhau.
18.3.3. Chọn buret thích hợp, đổ đầy 0,01 mol/L dung dịch rượu KOH, đưa buret đến vị trên bộ chuẩn độ, đảm bảo rằng đầu buret được nhúng sâu khoảng 25 mm trong bình chứa chất lỏng chuẩn độ và chuẩn độ.
18.3.4. Khi chuẩn độ xong, tráng các điện cực và đầu buret bằng proran-2-ol, sau đó bằng nước. Nhúng các điện cực vào trong nước ít nhất 2 min trước khi bắt đầu lần chuẩn độ khác để phục hồi lớp gel nước của điện cực thủy tinh. Nhúng các điện cực vào propan-2-ol trước khi tiếp tục đo mẫu khác. Nếu thấy các điện cực bị bẩn thì xử lý như 8.1. Bảo quản điện cực theo 8.3.3.
18.3.5. Nhiều điểm uốn chuẩn độ được tìm thấy khi phân tích các axit hữu cơ của quá trình oxy hóa điêzen sinh học khi tồn trữ quá lâu. Vì vậy, thể tích chuẩn độ điểm cuối rõ ràng cuối cùng được dùng để tính tổng lượng axit.
19. Cách tính hoặc giải thích kết quả
19.1. Cách tính nồng độ dung dịch KOH, mol/L:
19.1.1. Cách tính nồng độ molar KOH, mol/L bằng thể tích mol/L của dung dịch KHP:
|
Nồng độ dung dịch KHP, mol/L = |
khối lượng KHP, g |
(9) |
|
204,23 * (tổng thể tích dung dịch KHP, L) |
|
Nồng độ molar KOH, mol/L = |
(2,00 mL dung dịch KHP)(nồng độ của dung dịch KHP, mol/L) |
(10) |
|
thể tích của KOH, mL |
19.1.2. Cách tính KOH mol/L bằng khối lượng mg/g của dung dịch KHP:
|
Nồng độ dung dịch KHP, mg/g = |
(khối lượng KHP, g) * 1000 |
(11) |
|
204,23 * (tổng khối lượng dung dịch KHP, L) |
|
Nồng độ molar KOH, mol/L = |
(khối lượng dung dịch KHP, g)(nồng độ của dung dịch KHP, mg/g) |
(12) |
|
thể tích của KOH, mL |
CHÚ THÍCH 20: Giá trị trung bình mol/L của ba phép xác định được sử dụng để xác định trị số axit. Giá trị trung bình được chấp nhận lệch ± 0,0005 M.
19.2. Tính trị số axit:
Trị số axit, mg KOH/g = (A – B) x M x 56,1/W (13)
trong đó
A là thể tích dung dịch KOH trong alcoho dùng để chuẩn độ mẫu đến khi kết thúc điểm uốn cuối cùng, tính bằng mililit;
B là thể tích tương ứng với A dùng để chuẩn độ mẫu trắng, tính bằng mililit;
M là nồng độ của dung dịch KOH trong alcoho, tính bằng mol trên lít;
W là khối lượng mẫu, tính bằng gam.
20. Kiểm soát chất lượng
20.1. Kiểm tra xác nhận khả năng áp dụng của phương pháp bằng cách phân tích mẫu kiểm soát chất lượng (QC), mẫu này phải đại diện cho các mẫu được phân tích điển hình.
CHÚ THÍCH 21: Bởi vì điêzen sinh học có thể bị thay đổi do thời gian lưu trữ kéo dài nên mẫu không phù hợp với mục đích này. Người phân tích có thể dùng dung dịch kali axit phtalat 0,01 M như là chuẩn kiểm soát chất lượng. Khi được sử dụng là chuẩn kiểm soát chất lượng (QC), dung dịch KHP sẽ là chất chỉ thị tốt khi chuẩn hóa chất chuẩn độ (0,01 M KOH trong IPA) là cần thiết. Không có dữ liệu về thời hạn sử dụng của dung dịch KHP. Có thể sử dụng các dung dịch tiêu chuẩn thương phẩm.
20.2. Trước khi tầm soát các quá trình đo, thí nghiệm viên cần xác định giá trị trung bình và các giới hạn kiểm soát của mẫu kiểm soát chất lượng (QC).
20.3. Ghi các kết quả QC và phân tích theo biểu đồ hoặc kỹ thuật thống kê tương đương để đảm bảo việc kiểm soát mang tính thống kê cho toàn bộ quá trình thử nghiệm. Các số liệu ngoài vùng kiểm soát phải được nghiên cứu kỹ để tìm các nguyên nhân chính. Các kết quả này có thể dùng khi hiệu chuẩn thiết bị, nhưng không nhất thiết.
20.4. Tần suất thử nghiệm QC phụ thuộc vào tầm quan trọng của chỉ tiêu chất lượng đang kiểm tra, độ ổn định của quy trình thử, và các yêu cầu của khách hàng. Thông thường, hàng ngày phân tích mẫu QC, nếu số lượng mẫu đem phân tích lớn thì tăng tần suất số phân tích mẫu kiểm soát chất lượng lên. Tuy nhiên, khi công bố là phép thử đang được tiến hành dưới điều kiện kiểm soát mang tính thống kê thì có thể giảm tần suất thử nghiệm. Độ chụm của mẫu kiểm soát chất lượng phải được kiểm tra theo độ chụm quy định trong phần độ chụm và độ chệch của tiêu chuẩn để đảm bảo chất lượng các số liệu.
20.5. Khuyến cáo tiêu chuẩn QC được phân tích thường xuyên tại mức nồng độ trong cùng một phạm vi như trong mẫu được phân tích. Nguồn cung cấp mẫu QC phải sẵn sàng đáp ứng cho thời gian sử dụng mẫu dự kiến, mẫu phải đảm bảo đồng nhất, ổn định dưới điều kiện đảm bảo quy định.
21. Báo cáo kết quả
21.1. Báo cáo trị số axit của điêzen sinh học và hỗn hợp chính xác đến 0,01 mg KOH/g mẫu [TCVN 6325 (ASTM D 664), Phương pháp thử B].
22. Độ chụm và độ chệch
22.1. Độ chụm của phương pháp thử này dựa trên nghiên cứu liên phòng thử nghiệm của ASTM D 664 tiến hành năm 2009. Bảy phòng thử nghiệm tham gia nghiên cứu này, tuy nhiên các kết quả từ một phòng thử nghiệm đã loại trừ các cách tính độ chụm. Mỗi một phòng thử nghiệm được yêu cầu báo cáo các kết quả thí nghiệm kép của 11 mẫu điêzen, hỗn hợp điêzen sinh học và mẫu trắng khác nhau. Từng “kết quả thử nghiệm” đã báo cáo đại diện cho phép xác định hoặc phép đo đơn lẻ. D2PP được sử dụng để phân tích dữ liệu nghiên cứu, các chi tiết được nêu trong báo cáo nghiên cứu ASTM RR: D02 – 1727.
22.1.1. Giới hạn độ lặp lại (r): hai kết quả thử nghiệm thu được trong cùng một phòng thử nghiệm được đánh giá là không tương đương nếu sự khác nhau giữa chúng lớn hơn giới hạn độ lặp lại, “r” là khoảng giới hạn độ lặp lại đại diện cho sự khác biệt tới hạn giữa hai kết quả thử của cùng nguyên, do cùng một thí nghiệm viên tiến hành sử dụng cùng một thiết bị trong cùng một ngày và cùng một phòng thử nghiệm. Các giới hạn độ lặp lại được ghi trong Bảng 2.
22.1.2. Giới hạn độ tái lập (R): hai kết quả thử nghiệm được đánh giá là không tương đương nếu sự khác nhau giữa chúng lớn hơn giới hạn độ tái lập, “R” là khoảng giới hạn độ tái lập đại diện cho sự khác biệt tới hạn giữa hai kết quả thử của cùng nguyên liệu, do các thí nghiệm viên khác nhau tiến hành sử dụng thiết bị khác nhau trong các phòng thử nghiệm khác nhau. Các giới hạn độ tái lập được ghi trong Bảng 2.
22.1.3. Các thuật ngữ (giới hạn độ lặp lại và giới hạn độ tái lập) được sử dụng như quy định trong ASTM E 177.
22.1.4. Bất kỳ sự đánh giá nào đều phải phù hợp với 22.1.1 và 22.1.2 với xác suất chính xác khoảng 95 %.
22.2. Độ chụm được xác định thông qua việc thống kê kiểm tra 138 kết quả, lấy từ 6 phòng thử nghiệm trong tổng số 11 mẫu hỗn hợp xăng khác nhau và một mẫu trắng.
22.3. Độ chệch – trong thời gian nghiên cứu, không có vật liệu chuẩn được chấp nhận phù hợp để xác định độ chệch cho phương pháp thử này, do vậy không có công bố về độ chệch.
Bảng 2 – Trị số axit của điêzen sinh họcA
|
Độ lặp lại = 0,264E-01 * X ^ 0,4 mg/kg KOH Độ tái lập = 0,177 * X ^ 0,4 mg/kg KOH |
|
A Bậc tự do của R nhỏ hơn 30 và lớn hơn 15. Các mẫu 4, 5, 6, 9 bị loại vì ở dưới mức giới hạn định lượng của phương pháp thử |
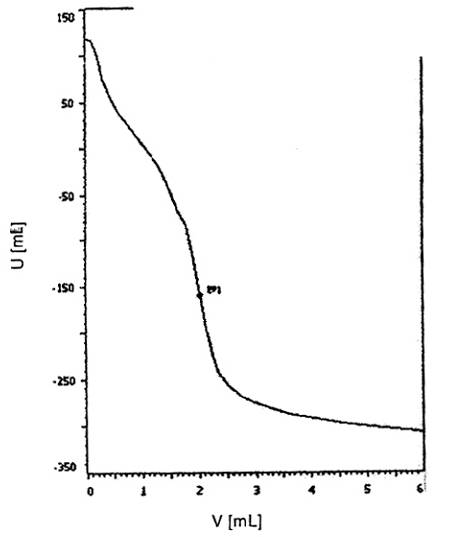
Hình 2 – Các đường cong chuẩn độ minh họa của mẫu điêzen sinh học
Phụ lục A
(tham khảo)
Kiểm tra tính năng của các điện cực
A.1. Phép thử bằng điện cực động học đo đáp ứng động học của điện cực. Có thể hiệu chuẩn các điện cực qua độ dốc chấp nhận được và các giá trị bị chặn chưa đáp ứng tốt để chuẩn độ. Tốc độ đáp ứng và độ ổn định rất quan trọng đối với điện cực chuẩn độ. Phép kiểm tra thủ công dưới đây có thể thực hiện bằng máy đo pH hoặc thiết bị chuẩn độ để có thể đọc liên tục các giá trị milivon.
A.1.2. Bản chất của phép kiểm tra này là kiểm tra khả năng của các điện cực khi nhúng trong dung dịch nước có chất đệm và đo thế sau 30 s và 60 s. Một điện cực nhanh sẽ đạt được điểm ổn định trong thời gian ít hơn 30 s và thay đổi ít trong khoảng từ 30 s đến 60 s. Khi kiểm tra, nếu cần có thể sử dụng các chất đệm pH 4, pH 7 và pH 11.
A.1.3. Cách tiến hành
A.1.3.1. Đặt thiết bị chuẩn độ hoặc máy đo pH để có thể đọc liên tục giá trị milivon. Cần trang bị dụng cụ khuấy dung dịch đệm có cùng tốc độ như khi chuẩn độ.
A.1.3.2. Để điện cực ổn định trong 1 min trong nước cất hoặc nước khử ion tương đương.
A.1.3.3. Lấy các điện cực ra khỏi nước và đặt trong dung dịch đệm pH 4. Bấm đồng hồ tại thời điểm khi dung dịch đệm chạm vào điện cực.
A.1.3.4. Ghi điện thế sau 30 s. Sau 30 s tiếp theo, ghi điện thế lần nữa. Sự chênh lệch giữa hai lần đo gọi là độ lệch.
A.1.3.5. Lặp lại quy trình đối với dung dịch đệm pH 7 và pH 11.
A.1.4. Tính độ lệch cho từng loại dung dịch đệm. Sự đáp ứng của điện cực có thể quy định như sau:
|
Độ lệch |
<> |
rất tốt |
|
1 < độ=””> |
<> |
tốt |
|
2 < độ=””> |
<> |
chấp nhận được |
|
3 < độ=””> |
<> |
cần xem xét |
|
4 < độ=””> |
không chấp nhận |
|
A.1.5. Sự chênh lệch giữa các thế ở 60 s đối với dung dịch đệm pH 4 và pH 7 phải lớn hơn 162 mV, hoặc 54 mV/pH. Các điện cực có độ dốc nhỏ hơn 54 mV/pH không nên dùng để chuẩn độ.
